فرط كوليسترول الدم العائلي
فَرْطُ كوليسترول الدَّم العائِلِيّ*[1][2][3] (بالإنجليزية: Familial hypercholesterolemia) ويُدعى اختصارًا FH[3]، هوَ اضطرابٌ جيني، يتميز بارتفاعِ مستوياتِ الكوليسترول في الدم وتحديدًا ارتفاعُ مستوياتِ البروتين الدُهني مُنخفض الكثافة (الكوليسترول السيء)ب بشكلٍ عالٍ، مما يُساعدُ على الظهورِ المُبكر للأمراضِ القلبية الوعائية. نظرًا للاختلافِ الطفيف بين الأفراد في كيميائية الجِسم الحيوية التابعة لفرطِ كوليسترول الدم العائلي؛ فإنَّ مستوياتِ الكوليسترول المرتفعة تكونُ أقل استجابةً لأساليبِ ضبطِ الكوليسترول، والتي عادةً ما تكونُ أكثرَ فعاليةً في الأشخاص غير المُصابين بفرط كوليسترول الدم العائلي، ومن هذه الأساليب، تعديلُ النظام الغذائي أو أقراصِ الستاتين، ولكن على الرغمِ من ذلك، فإنَّ العلاجِ المُتضمن لجرعاتٍ عالية من الستاتين، عادةً ما يكون ناجعًا.
| فرط كوليسترول الدم العائلي | |
|---|---|
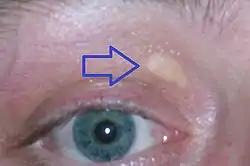 لويحة صَفراء جفنية، هي بقعٌ صفراء تتكونُ من ترسباتِ الكوليسترول أعلى الجفن، ويزدادُ ظهورها لدى الأشخاص المُصابين بفرطِ كوليسترول الدَّم العائِلِي لويحة صَفراء جفنية، هي بقعٌ صفراء تتكونُ من ترسباتِ الكوليسترول أعلى الجفن، ويزدادُ ظهورها لدى الأشخاص المُصابين بفرطِ كوليسترول الدَّم العائِلِي | |
| معلومات عامة | |
| الاختصاص | علم الغدد الصم |
| من أنواع | فرط شحميات الدم العائلي ، وفرط كوليسترول الدم |
| الإدارة | |
| أدوية | |
يُصنف فرط كوليسترول الدم العائلي على أنهُ النمط الأول من النوعُ الثاني من اختلالِ دهونِ الدم العائلي، حيثُ يوجدُ خمسُ أنواعٍ من اختلالِ دهونِ الدم العائلي (لا تتضمن الأنواع الفرعية)،[4][5] ويُقسم كل نوعٍ وفقًا للملف الليبيدي المُتغير والاضطرابات الجينية، فمثلًا، يتصفُ النوع الثاني بارتفاعِ مستوياتِ البروتين الدُهني مُنخفض الكثافة، والذي عادةً ما يكونُ بسبب عيبٍ في مستقبل هذا البروتين الدهني، أما الأنواع الأخرى، فتتضمنُ عيبًا في أيضٍ كلٍ من الكيلومكرون وثلاثي الغليسريد والجُزيئات الأخرى المحتوية على الكوليسترول مثلَ، البروتينات الدهنية متوسطة الكثافة والبروتينات الدهنية منخفضة الكثافة جدًا.[6]
شَخصٌ واحد في كل 300 إلى 500 شَخص لديهِ طفراتٌ في جين مستقبل البروتين الدهني منخفض الكثافة (LDLR) والذي يُشفر بروتين هذا المُستقبل، والذي عادةً ما يُزيل البروتين الدهني مُنخفض الكثافة من الدورة الدموية، أو يُزيل صميم البروتين الدهني بي، والذي يُعتبر جزءًا من البروتين الدهني مُنخفض الكثافة، والذي يرتبطُ مع المُستقبل، أما الطفراتُ في الجيناتِ الأخرى فهي نادرة الحدوث.[7] الأشخاص الذين يمتلكونُ نسخةً واحدة شاذة (مُتغايري الزيجوت) من الجين، قد تتطورُ لديهم أمراضٌ قلبية وعائية مُبكرة ما بين سن 30 إلى 40 عامًا، أما من يمتلكون نُسختين شاذتين (متماثلي الزيجوت) فإنهُ قد يحدثُ لديهم أمراضٌ قلبية وعائية شديدة في مرحلةِ الطفولة. يُعتبر فرط كوليسترول الدم العائلي مُتغاير الزيجوت من الاضطرابات الجينية الشائعة، ويورثُ بنمطِ الوراثة الجسمية السائدة، ويحدثُ بنسبة 1:500 شخص في مُعظم الدول،[8] أما النوع مُتماثل الزيجوت فهو نادرُ الحدوث، ولكن يحدث في حوالي 1 لكل مليون ولادة.[8][9]
يُعالج فرط كوليسترول الدم العائلي مُتغاير الزيجوت عادةً باستخدامِ الستاتين أو منحيات حامض الصفراء أو الأدوية الأُخرى الخافِضة لشحمياتِ الدِم والتي تعملُ على تقليل مستوياتِ الكوليسترول، كما يُقدم للحالات الجديدة استشارة وراثية، أما فرط كوليسترول الدم العائلي مُتماثل الزيجوت، فعادةً لا يستجيبُ للعلاجِ الطبي، وقد يتطلبُ علاجاتٍ أُخرى، مثل فِصادَة البروتين الدهني مُنخفض الكثافة (إزالةُ البروتين الدهني منخفض الكثافة بطريقةٍ تُشبهُ الغسيل الكلوي)، أو في بعضِ الأحيان قد يُضطرُ إلى زراعة الكبد.[8]
الأعراض والعلامات
العلامات الجسمية
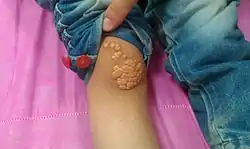
طبيعيًا، لا يُسبب ارتفاع مستويات الكوليسترول أيَ أعراضٍ، ولكن يُمكن ملاحظةُ ترسباتٍ صفراءَ من الدُهون الغَنية بالكوليسترول في أماكنَ مُختلفة من الجسم، مثلً حولَ الجفون والتي تُعرف باسم اللويحات الصفراء الجفنية، وهي تجمعاتٌ صفراء من الكوليسترول تحت الجلد، وهي غيرُ مؤذيةٍ للجلد وليست مؤلمة.[10] أيضًا يُمكن أن تظهر هذه الترسبات في الحافةِ الخارجية من القزحية وتعرف باسم قوس الشيخوخة، أو قد تظهر في أوتارِ اليدين والمرفقين والركبتين والقدمين، وخاصةً وتر أخيل، وتُعرف الحالة باسم الورم الأصفر الوتري.[8][11]
.jpg.webp)
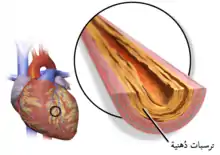
الأمراض القلبية الوعائية
تَحدث الأمراضُ القلبية الوعائية بسببِ الترسُبِ السريع للكوليسترول في جُدرانِ الشرايين، مؤديةً إلى تصلبٍ عصيدي. المُشكلة الأكثر شيوعًا في فرط كوليسترول الدم العائلي هي حدوثُ مرض الشريان التاجي (تصلبُ الشرايين التاجية التي تُغذي القلب) في فئاتٍ عمرية أصغرُ مما هو مُتوقعٌ في المجتمع. قد يؤدي هذا المرض إلى حدوثِ ذبحةٍ صدرية (ألم أو ضيق في الصدر عندَ بذلٍ مجهودٍ ما) أو نوبةٍ قلبية. قد تتأثرُ شرايين الدماغ أيضًا، ولكن بدرجةٍ أقلُ شيوعًا، وتأثُرها قد يؤدي إلى نوباتٍ إقفاريةٍ عابرة (نوباتٍ قصيرة من ضعفٍ جانبٍ واحدٍ من الجسم أو عدمِ القدرة على الكلام) أو سكتةٍ أحيانًا.[9]
يحدثُ مرض انسداد الشريان المُحيطي بشكلٍ رئيسيٍ في المُدخنين المُصابين بفرط كوليسترول الدم العائلي، وهذا قد يؤدي إلى ألمٍ في عضلاتٍ بطن الساق أثناءَ المشي، ويذهبُ مع الراحة، ويُعرف باسم العرج المُتقطع، كما قد يؤدي إلى مشاكلَ أُخرى ناجمة عن انخفاضِ تدفقِ الدمِ إلى القدمين، مثل الغنغرينا.[9] يزدادُ خطرُ الإصابة بالتصلبُ العصيدي مع ازديادِ العمرِ ولدى المُدخنين، ومرضى السكري وارتفاع ضغط الدم، وفي حال وجودِ تاريخٍ عائلي للإصابةِ بالأمراض القلبية الوعائية.[8][12]
التشخيص
| مَقاييس التشخيص لفرط كوليسترول الدم العائلي مُتغاير الزيجوت المُحتمل
(النوعية 98%)[13] | ||||||
|---|---|---|---|---|---|---|
| قرابة من الدرجة الأولى | جميع السُكان | |||||
| العمر | الكوليسترول | مغ/دل | ممل/ل | مغ/دل | ممل/ل | |
| < 18 | الإجمالي | > 220 | > 5.7 | > 270 | > 7.0 | |
| LDL-C | > 155 | > 4.0 | > 200 | > 5.2 | ||
| 20–29 | الإجمالي | > 240 | > 6.2 | > 290 | > 7.5 | |
| LDL-C | > 170 | > 4.4 | > 220 | > 5.7 | ||
| 30–39 | الإجمالي | > 270 | > 7.0 | > 340 | > 8.8 | |
| LDL-C | > 190 | > 5.0 | > 240 | > 6.2 | ||
| ≥ 40 | الإجمالي | > 290 | > 7.5 | > 360 | > 9.3 | |
| LDL-C | > 205 | > 5.3 | > 260 | > 6.7 | ||
| ||||||
حوالي 85% من الأشخاصِ المُصابين بفرط كوليسترول الدم العائلي لم يُشخصوا وبالتالي لا يتلقونَ علاجاتِ خفضِ الدهون.[14] قد تُساعدُ نتائجُ الفحص الجِسمي الطَبيبَ على تشخيص فرط كوليسترول الدم العائلي، حيثُ يحدث الورم الأصفر الوتري في حوالي 20-40% من الحالات، ويُعتبرَ واصمًا مرضيًا للحالة،[14] وأيضًا قد يظهرُ لدى المُصابين لويحاتٌ صَفراء أو قوسُ الشيخوخة. جميع هذه العلامات الجسمية تُدعم التشخيص بفرط كوليسترول الدم العائلي، ولكنها غيرُ خاصةٍ لفرط كوليسترول الدم العائلي فقط.[14]
قياسات الليبيدات
يُمكن اعتبارُ تحديد مُستويات الكوليسترول جزءًا من فحصِ التأمين الصحي أو الصحة المهنية، وذلكَ في حال لوحِظت العلاماتُ الجسمية الخارجية مثل اللويحات الصفراء وقوس الشيخوخة وغيرها، أو في حالِ ظهورِ أعراض الأمراض القلبية الوعائية، أو وجودِ تاريخٍ لفردٍ من العائلةِ مُصابٌ بفرط كوليسترول الدم العائلي. عادةً ما يُوجد نمطٌ مُتوافقٌ مع فرط بروتينات الدم الشحمية من النمط IIa وذلك وفقَ تصنيف فريدريكسون، وهو: ارتفاعُ مستوى الكوليسترول الكُلي (أو الإجمالي)، وزيادةٌ ملحوظة في مستوى البروتين الدهني مُنخفض الكثافة، ومستوًى طبيعي من البروتين الدُهني مرتفع الكثافة ومن ثلاثي الغليسريد. يبلغ مستوى الكوليسترول الكُلي حوالي 350-550 مغ/دل في النوعِ مُتغاير الزيجوت، أما النوع مُتماثل الزيجوت، فيبلغُ مستوى الكوليسترول الكُلي فيه حوالي 650-1000 مغ/دل.[14] عادةً ما يكون البروتين الدهني مُنخفض الكثافة أعلى من 75 مئين؛ أي أنَّ 75% من السُكان الأصِحاء سيكون لديهم مستوًى مُنخض من البروتين الدهني مُنخفض الكثافة.[8] يُمكن أن تكون مُستويات الكوليسترول عالية كثيرًا في الأشخاص الذين يُعانون من السمنة.[9]
تحليل الطفرة
اعتمادًا على عزلِ البروتين الدهني منخفض الكثافة وعلى المقاييسِ السريرية (والتي تختلفُ بين الدُول)، يُمكن إجراء الاختبارات الجينية لطفراتِ مستقبل البروتين الدهني منخفض الكثافة وطفراتِ صميم البروتين الدهني بي.[15] عادةً تُكتشفُ الطفرات في 50-80% من الحالات، أما من لم تظهر لديهم الطفرات، فعادةً يكون لديهم مستوياتٌ مرتفعة من ثلاثي الغليسريد، وقد يكونُ لديهم أسبابٌ أخرى لارتفاع نسبة الكوليسترول في الدم، مثل فرط دهنيات الدم المركب والناتجِ عن المتلازمة الأيضية.[15]
على الرغم من ذلك، فإنه ليست جميعُ الطفراتِ المعروفة للجين المُشفرِ لمستقبل البروتين الدهني منخفض الكثافة تُسببُ آثارًا مؤذيةً على مستويات كوليسترول الأفراد، حيثُ توجدُ طفراتٌ يُمكن وصفها بالمُفيدة في ضبطِ وتعديل مستويات الكوليسترول في البلازما، ومن الأمثلة عليها طفرةُ استبدال الفالين بواحدٍ من الألانين في موضعِ 578 لمستقبل البروتين الدهني منخفض الكثافة (Val578Ala)، والتي تحدثُ في السردينيين، مما يؤدي إلى تقليل مستويات كوليسترول البروتين الدهني منخفض الكثافة ومستوى الكوليسترول الكُلي في حاملي المرض.[16]
التشخيص التفريقي
يجبُ تمييزُ فرط كوليسترول الدم العائلي عن فرط دُهنيات الدم المركب وفرط كوليسترول الدم متعدد الجينات، حيثُ أنَّ مستوى الليبيدات ووجودُ الورم الأصفر يُساعد في تأكيد التشخيصِ بفرط كوليسترول الدم العائلي.[17] حالات وجود السيتوستيرول في الدم وداء الأورام الصفر المنتشرة الدماغية الوترية، وهي حالاتٌ طبية نادرة، قد تترافق مع الورم الأصفر وتصلبٍ عصيدي المُبكر، كما أنَّ داء الأورام الصُفر المُنتشرة الدماغية الوترية قد يُتضمنُ أعراضًا عصبية أو نفسية، وعتامة عدسة العين وإسهال واضطراباتٍ هيكلية.[17]
علم الوراثة
تُعتبر طفراتُ جين مستقبل البرتين الدهني منخفض الكثافة (LDLR) من أكثرِ العيوبِ الوراثية شيوعًا في فرط كوليسترول الدم العائلي، حيثُ أنَّ انتشارها 1 لكل 500 شخص، ويعتمدُ الأمر على السكان. ومن الطفراتِ الأخرى، طفراتُ صميم البروتين الدهني بي (ApoB)، وانتشارها 1 لكل 1000، وطفراتُ جين النوعِ التاسع سبتيليزين/كيكسين كونفيرتاز طليعة البروتين (PCSK9) وانتشاره أقلُ من 1 لكل 2500، وقد تحدث أيضًا طفراتُ جين البروتين الأول المكيِّف لمستقبل البرتين الدهني منخفض الكثافة (LDLRAP1). مرضَ وجود السيتوستيرول في الدم والذي يمتلكُ تشابهاتٍ عديدة مع فرط كوليسترول الدم العائلي، ويمتلكُ ميزاتِ تراكم الكوليسترول في الأنسجة، فإنه يحدثُ نتيجةً لطفراتِ جين الأدينوسين ثلاثي الفوسفات المرتبط بحافظة العضو الثامن من فصيلة جي (ABCG8) وجين المرتبطة بحافظة العضو الخامس من فصيلة جي (ABCG5).[8]
مستقبل البروتين الدهني منخفض الكثافة
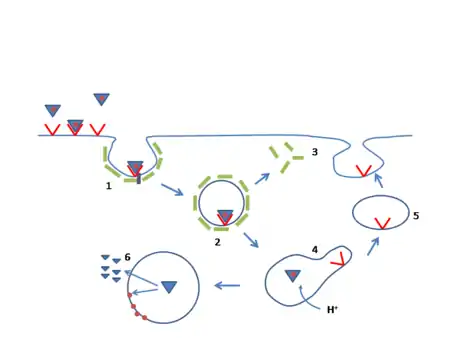
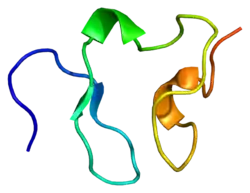
يقعُ جين مستقبل البروتين الدهني منخفض الكثافة على الذراعِ القصيرة للصبغي 19 (19p13.1-13.3)،[14] ويتألفُ الجين من 18 إكسون ويتسع إلى 45 كيلو زوج قاعدي (كيلوبيز)، كما أنَّ البروتين المُنتج من الجين في شكلهِ الناضج يحتوي على 839 حمض أميني. تُسبب نُسخة واحدة شاذة (مُتغاير الزيجوت) من فرط كوليسترول الدم العائلي أمراضًا قلبية وعائية في سن الخمسين لدى حوالي 40% من الحالات، ووجودُ نسختين شاذتين (مُتماثل الزيجوت) تُسبب تُسارعًا في حدوثِ التصلب العصيدي في مرحلةِ الطفولة، ويشملُ أيضًا ما يترتب عليه من مُضاعفاتٍ. تَتَناسَبُ مُستويات البروتين الدهني منخفض الكثافة في البلازما عكسيًا مع نشاط مستقبل البروتين الدهني منخفض الكثافة. يبلغَ نشاطُ مستقبل البروتين الدهني منخفض الكثافة في متماثلِ الزيجوت أقل من 2%، أما في مُتغاير الزيجوت فهُناك عيبٌ في معالجة البروتين الدهني منخفض الكثافة ويبلغُ نشاط المستقبل ما بين 2-25%، وهذا يعتمدُ على طبيعة الطفرة. هُناك أكثر من 1000 طفرة مُختلفة معروفة.[8]
هُناك خمسُ أصنافٍ رئيسية من فرط كوليسترول الدم العائلي نتيجةً لطفرات البروتين الدهني منخفض الكثافة:[18]
- الصنفُ الأول: لا يُصنع مستقبل البروتين الدهني منخفض الكثافة نهائيًا.
- الصنفُ الثاني: لا يُنقلُ مستقبل البروتين الدهني منخفض الكثافة جيدًا من الشبكة الإندوبلازمية إلى جهاز غولجي الذي يُعبر عنهُ على سطحِ الخلية.
- الصنفُ الثالث: لا يرتبطُ المستقبل جيدًا مع البروتين الدهني منخفض الكثافة على سطحِ الخلية؛ بسببِ عيبٍ إما في صميم البروتين الدهني بي100 (R3500Q) أو في المُستقبل نفسه.
- الصنفُ الرابع: لا يتكون العُنقود البروتين الدُهني منخفض الكثافة في وهداتِج الكلاثرين المُغطاة، الضرورية لحصولِ الإدخال الخلوي المتواسط بالمستقبل (خطوة المسار الثانية).
- الصنفُ الخامس: لا يُعادُ استخدام المستقبل مرةً أخرى في سطح الخلية (خطوة المسار الخامسة).
صميم البروتين الدهني بي
يُعتبر الشكل (ApoB100) من صميم البروتين الدهني بي، الجزء البروتيني في الجسيم البروتيني الدهني أو الصميم البروتين الدهني الرئيسي. يقعُ جين صميم البروتين الدهني بي على الصبغي الثاني (2p24-p23)، وطولهُ ما بين 21.08 و21.12 ميجا زوج قاعدي (ميجابيز).[19] عادةً يرتبطُ فرط كوليسترول الدم العائلي مع طفرةٍ في R3500Q، والتي تؤدي إلى استبدالِ الأرجنين بالجلوتامين في الموضع 3500، حيثُ تقع الطفرة على جزءٍ من البروتين الذي عادةً ما يرتبطُ بمستقبل البروتين الدهني مُنخفض الكثافة، ويقلُ هذا الارتباط نتيجةً للفطرة. كما هوَ الحال في جين مستقبل البروتين الدهني مُنخفض الكثافة، فإنَّ عدد النسخات الشاذة يُحدد شدة فرط كوليسترول الدم.[8][19]
PCSK9
ترتبطُ طفراتُ النوعِ التاسع سبتيليزين/كيكسين كونفيرتاز طليعة البروتين مع فرط كوليسترول الدم العائلي الجسمي السائد وراثيًا (أي يحتاجُ نسخةً شاذة واحدة) وذلك حسب تقرير عام 2003.[8][20] يقعُ هذا الجين على الصبغي الأول (1p34.1-p32)، ويُشَفِر بروتينًا طولهُ 666 حمضًا أمينيًا والذي يُعبرُ عنه في الكبد. اقتُرحَ أنَّ PCSK9 يُسبب فرط كوليسترول الدم العائلي رئيسيًا عبر تقليل عدد مستقبلات البروتين الدهني منخفض الكثافة في خلايا الكبد.[21]
LDLRAP1
أُبلغ عن اضطرابات البروتين الأول الملئم لمستقبل البرتين الدهني منخفض الكثافة (LDLRAP1) للمرةِ الأولى في عائلةٍ عام 1973،[22] وعلى عكسِ الطفراتِ الأخرى، فإنهُ يجب وجود نسختين شاذتين من الجين لحصولِ فرط كوليسترول الدم العائلي (وراثة جسمية متنحية). تؤدي الطفرات في البروتين إلى إنتاجِ بروتين قصير، كما أنَّ وظيفته الحَقيقة غير واضحة حتى الآن، لكنه يبدو أنهُ يلعبُ دورًا في العلاقة ما بينَ مستقبل البروتين الدهني منخفض الكثافة ووهداتِ الكلاثرين المُغطاة. المُصابين بفرط كوليسترول الدم الجسمي المُتنحي يميلون لحدوثِ أمراضِ شديدة لديهم أكثر من مُتغايري زيجوت مستقبل البروتين الدهني منخفض الكثافة ولكنَّ أقلُ من مُتماثلي زيجوت المستقبل.[8]
جيناتٌ أخرى
نُشرت دراسةٌ حديثة في دورية نيتشر جيناتكس (Nature Genetics)، ذُكرَ فيها أنَّ طفرة β039 ترتبطُ مع انخفاضِ مستويات الكوليسترول الكُلي وكوليسترول البروتين الدهني مُنخفض الكثافة في البلازما.[16] الأمرُ الغريب، أنَّ هذه الطفرة تقع على جين بيتا غلوبين (HBB) والذي يُشَفِرُ سلسلة بيتا في الغلوبين، وهو السبب الرئيسي لحدوثِ مرضِ بيتا ثلاسيميا (يُورث بنمطِ الوراثة الجسمية السائدة) في سكانُ جزيرة سردينيا.[16]
الفيزيولوجيا المَرضيّة
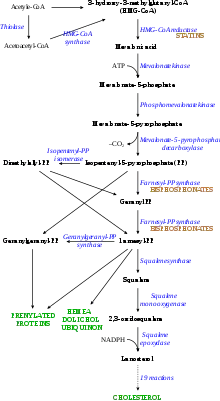
 مقالات مفصلة: مستقبل بروتين دهني منخفض الكثافة
مقالات مفصلة: مستقبل بروتين دهني منخفض الكثافة- تصلب عصيدي
طبيعيًا، يَدورُ كوليسترول البروتين الدهني مُنخفض الكثافة في الجسم لمدة يومين ونصف اليوم، وفيما بعد يرتبطُ جزء صميم البروتين الدهني بي الخاص بكوليسترول البروتين الدهني مُنخفض الكثافة معَ مستقبل البروتين الدهني مُنخفض الكثافة الموجود على خلايا الكبد، محفزًا امتصاصه وهضمه.[14] ينتجُ عن هذه العملية، إزالةُ البروتين الدهني منخفض الكثافة من الدورة الدموية. يُثبَطُ تَخليق الكبد للكوليسترول في مسار الميفالونات (مسار مختزلة 3-هيدروكسي-3-ميثيل جلوتاريل تميم الإنزيم أ).[23] تتأثر وظيفة مستقبل البروتين الدهني مُنخفض الكثافة في فرط كوليسترول الدم العائلي، حيث قد تنخفض أو تختفي تمامًا،[14] كما أنَّ البروتين الدهني مُنخفض الكثافة يدورُ في الجسم لمدةٍ متوسطها أربعُ أيامٍ ونصف اليوم، مما يؤدي إلى زيادةٍ كبيرة في مستوى كوليسترول البروتين الدهني مُنخفض الكثافة في الدم مع مستوياتٍ طبيعية للبروتينات الدُهنية الأخرى.[9] في طفراتِ صميم البروتين الدهني بي، يؤدي انخفاضُ ارتباط جزئيات البروتين الدهني مُنخفض الكثافة مع المُستَقبِل إلى زيادةِ مستوى كوليسترول البروتين الدهني مُنخفض الكثافة. من غيرِ المعروف كيف تُسبب طفراتُ PCSK9 وLDLRAP1 اختلالًا في المستقبل.[8]
على الرغمِ من أنَّ التصلب العصيدي يحدثُ في جميع الأشخاصِ بدرجةٍ مُعينة، إلا أنَّ المُصابين بفرط كوليسترول الدم العائلي يتسارعُ لديهم حدوثُ هذا التصلب؛ بسببِ مستوى البروتين الدهني مُنخفض الكثافة المُرتفع. تعتمد درجةُ التصلب العصيدي تقريبًا على عددِ مستقبلات البروتين الدهني منخفض الكثافة التي لا زالَ يُعبر عنها، كما ويعتمدُ على وظائفِ هذه المستقبلات. في العديد من حالاتِ فرط كوليسترول الدم العائلي مُتغاير الزيجوت، تكونُ وظيفة المستقبلات ضعيفةٍ قليلًا، ومستويات البروتين الدهني مُنخفض الكثافة مُنخفضة نسبيًا، أما في حالات متماثلي الزيجوت، فإنَّ لا يُعبر عن المستقبل نهائيًا.[8]
تُشير بعضُ دراسات الأترابِ حول فرط كوليسترول الدم العائلي إلى وجودِ عوامل خطرٍ إضافية تلعبُ دورًا عندما يُصاب الشخص بالتصلب العصيدي،[24][25] إضافةً إلى ذلك، توجدُ عواملُ خطرٍ تقليدية مثل التدخين وارتفاعِ ضغط الدم والسكري، وأظهرت دراساتٌ جينية أنَّ اضطرابًا شائع في جين الثرومبين (G20210A) يزيد من خطرِ حدوث الأمراض القلبية الوعائية في مرضى فرط كوليسترول الدم العائلي.[26] وجدت دراساتٌ مُتعددة أنَّ ارتفاعَ مستوى البروتين الدهني(a) يُشكلُ عاملَ خطرٍ إضافيًا لحدوث مرض القلب الإقفاري.[27][28] كما أنهُ يزدادُ الخطر في الأشخاص الذين يمتلكون أنماطًا جينية مُحددة من الإنزيم المحول للأنجيوتنسين.[29]
الكشف
يُعتبر فحصُ (كشف) أفرادِ العائلة المعروفٌ إصابتهم بفرط كوليسترول الدم العائلي ذا تكلفةٍ فعالة.[30] في عام 2001، اقتُرح استخدامُ إستراتيجياتٍ أُخرى مثل إجراءِ كشفٍ شامل في سن 16 عامًا،[31][32] ولكنهُ قد يكونُ أقلَ فعاليةً من حيث التكلفة على المدى القصير.[33] يؤدي الكشفُ في سنٍ أقلُ من 16 عامًا إلى معدلٍ مرتفع غيرُ مقبولٍ من النتائج الإيجابية الزائفة.[9]
وجدَ تحليلٌ تلوي أجريَ في عام 2007 أنَّ "الإستراتيجية المُقترحة للكشف على الأطفال والآباء لفرط كوليسترول الدم العائلي، قد يكونُ لها تأثيرٌ هامٌ في منع الآثار الطبية لهذا الاضطراب في جيلين ضمنَ وقتٍ واحد"،[34] كما أنَّ "استخدام الكوليسترول الكُلي لوحدهِ قد يُميز بين الأشخاص الذين يعانون من فرط كوليسترول الدم العائلي والذين لا يعانون منه بين سن سنةٍ إلى 9 سنواتٍ".[34][35]
اقتُرحَ أن يُكشف على الأطفالِ المُتهادين، وفي عام 2016 نُشرت نتائجُ تجربةٍ على 10,000 طفلٍ في سنِ عامٍ واحد، ولكن هُناك حاجةٌ للعلم على إيجاد ما إذا كانَ الكشفُ عليهم فعالًا من حيث التكلفة ومقبولًا للأُسر.[36][37]
العلاج
متغايرو الزيجوت
غالبًا يُعالج فرط كوليسترول الدم العائلي بواسطة الستاتين،[14] والذي يعملُ على تثبيطِ إنزيم مختزلة 3-هيدروكسي-3-ميثيل جلوتاريل تميم الإنزيم أ (HMG-CoA-reductase) في الكبد، وردًا على ذلك، يقومُ الكبد بإنتاجِ مستقبلات البروتين الدهني مُنخفض الكثافة، والتي تُزيل البروتين الدُهني منخفض الكثافة من الدم. يُقلل الستاتين مستوياتِ الكوليسترول والبروتين الدهني منخفض الكثافة بفعاليةٍ، ومع ذلك، فإنَّ إضافةً أدويةٍ أخرى للعلاج قد يكونُ مطلوبًا أحيانًا، ومنها منحيات حامض الصفراء (كوليسترامين أو كوليستيبول) ومُستحضراتُ حمض النيكوتينك أو الفايبرات.[8] أيضًا يجبُ ضبطُ عواملِ الخطرِ الأخرى للأمراضِ القلبية الوعائية، حيثُ يبقى الخطرُ مرتفعًا إلى حدٍ ما حتى لو ضُبطت مستوياتُ الكوليسترول. تُوصي التوجيهاتُ المهنية، بأنَّ اتخاذ القرار لمعالجةِ شخصٍ مُصابٍ بفرط كوليسترول الدم العائلي بواسطة الستاتين يجب أن لا يعتمدَ على الأدواتِ المُعتادة للتنبؤ بالمخاطر (مثل تلكَ المُستمدة من دراسة فرامنغهام للقلب)؛ وذلك لأنه من المُحتمل أن لا يأخذ بعينِ الاعتبار خطرَ حدوث الأمراض القلبية الوعائية. المُصاب بفرط كوليسترول الدم العائلي يكونُ عكس أفرادِ المجتمع الآخرين، حيثُ يمتلكُ مستوياتٍ عالية من الكوليسترول منذُ الودلاة، مما قد يزيدُ من خطرها النسبي.[38] قبل دخولِ الستاتين حيزَ الاستخدام، كانت تستعمل أدويةٌ أخرى لخفضِ مستويات كوليسترول البروتين الدهني منخفض الكثافة، وهي الكلوفايبرات (فايبرات قديم، يُسبب حصواتٍ صفراوية) والبروبوكول (خاصةً عندَ وجودِ ورمٍ أصفر كبير) والثيروكسين.
تُعتبر إضافةُ الإزيتمايب للعلاجِ مثيرةً للجدل، حيثُ يثبطُ امتصاص الكوليسترول في القناةِ الهضمية، وعلى الرغمِ من أنهُ يقللُ كوليسترول البروتين الدهني مُنخفض الكثافة إلا أنه لم يظهرُ أيُ تحسنٍ في علامة التصلب العصيدي المعروفة باسم سماكة الغلالة الباطانة-الوسطانية، ومن غيرِ المعروف ما إذا كان هذا الأمر يعني أنَّ الإزيتمايب ليسَ له فائدةٌ عامة.[39]
لا تُوجد دراسات تداخلية تُظهر مباشرةً فوائدِ انخفاض الكوليسترول على الوفاة لدى مرضى فرط كوليسترول الدم العائلي. بدلًا من ذلك، استُمدت أدلةٌ على الفائدة من تجاربَ متعددة أُجريت على الأشخاص الذي يعانون من فرط كوليسترول الدم متعدد الجينات (حيثُ تلعب الوراثة فيه دورًا أقل). على الرغم من ذلك، في عام 1999 أظهرت دراسةٌ رصدية على سجلٍ بريطانيٍ كبير، أنهُ قد بدأ مُعدل الوفيات بين مُصابي فرط كوليسترول الدم العائلي بالتحسُن في أوائل التسعينات عندما أُدخلَ الستاتين.[40]
اقترحت دراسةٌ أترابية أنَّ علاجَ فرط كوليسترول الدم العائلي بالستاتين يؤدي إلى انخفاضٍ بنسبة 48% في الوفاةِ الناجمة عن مرضِ القلب التاجي إلى نقطةٍ ينعدمُ الموت فيها بسبب مرض القلب التاجي مقارنةً مع باقي السُكان، ولكن إذا كانَ الشخص يعاني بالفعلِ من مرض القلب التاجي فإنَّ الانخفاضَ يكونُ بنسبةِ 25%. تؤكد النتائج على ضرورةِ التحديدِ المُبكر لفرط كوليسترول الدم العائلي وعلاجه بالستاتين.[41]
الأليروكوماب والإيفولوكوماب، هما نوعان من الأجسامِ المُضادة أحادية النسيلة ضد النوعِ التاسع سبتيليزين/كيكسين كونفيرتاز طليعة البروتين (PCSK9)، ويوصفانِ خصوصًا كإضافةٍ للنظام الغذائي وللعلاج المُقاوم الأقصى للستاتين في البالغين المُصابين بفرط كوليسترول الدم العائلي مُتغاير الزيجوت، حيثُ يحتاجون إلى خفضٍ إضافي في كوليسترول البروتين الدهني منخفض الكثافة.[42]
متماثلو الزيجوت
علاجُ مُتماثلي الزيجوت أصعبُ من متغايري الزيجوت، حيثُ أنَّ مستقبلاتِ البروتين الدهني مُنخفض الكثافة تعملُ بشكلٍ طفيف أو رُبما لا تعمل. يحتاجُ مُتماثلي الزيجوت إلى جرعاتٍ عالية من الستاتين، وتكونُ في تركيبةٍ مع أدويةٍ أُخرى، حيثُ تساعد قليلًا على تحسين مستويات الليبيدات.[43] إذا لم ينجحِ العلاج بالأدوية على تخفيض مستويات الكوليسترول، فيُمكن استعمالُ فصادة البروتين الدهني منخفض الكثافة، والتي ترشح البروتين الدهني منخفض الكثافة من مجرى الدم، في عمليةٍ مُشابهةٍ للغسيل الكلوي.[8] الحالاتُ الشديدة من فرط كوليسترول الدم العائلي تُوضع لزراعة الكبد، حيثُ يُوفرُ كبد بمستقبلاتِ وظيفية للبروتين الدهني منخفض الكثافة، وهذا يؤدي إلى تحسنٍ سريع في مستويات الكوليسترول، ولكنَه في خطرٍ من حدوث مضاعفاتِ زراعة الأعضاء الصلبة (مثل رفضِ الزراعة والعدوى والآثار الجانبية للأدوية الضرورية لكبتِ الرفض).[44][45] تتضمن التقنياتُ الجراحية الأخرى، جراحة تجاوز الأمعاء اللفائفية الجزئية، والتي فيها يُتجاوز جزءٌ من الأمعاء الدقيقة لتقليلِ امتصاص العناصر الغذائية والتي تتضمن الكوليسترول، وأيضًا يُمكن إجراء جراحة التحويلة البابية الأجوفية، والتي فيها يُربط الوريد البابي مع الوريد الأجوف مما يسمحُ للدم مع العناصر الغذائية في الأمعاء بتجاوزِ الكبد.[46][47][48]
اللوميتابيد هو مُثبطٌ للبروتين الناقل لثلاثي الغليسريد الصغروري،[49] وقد وافقت عليه إدارة الغذاء والدواء الأمريكية في ديسمبر 2012، وذلك ليُستَعمل كدواءٍ يتيم في علاجِ فرط كوليسترول الدم العائلي متماثل الزيجوت.[50] في يناير 2013، وافقت إدارة الغذاء والدواء على دواءِ ميبومرسين، والذي يثبطُ عملَ جين صميم البروتين الدهني بي، مما يساعدُ في علاجِ فرط كوليسترول الدم العائلي متماثل الزيجوت.[51][52] يُعتبر العلاج الجيني البديل المُستقبلي المُحتمل.[53]
الأطفال
بما أنَّ فرط كوليسترول الدم العائلي قد يحدثُ منذُ الولادة وقد تبدأ علاماتُ التصلب العصيدي بالظهورِ مبكرًا،[54] فمن الضروري أحيانًا علاجُ اليافِعين وحتى المراهقين بأدويةٍ وعواملَ طُورتَ أصلًا لعلاجِ البالغين. يُفضل العديدُ من الأطباء استخدام منحيات حامض الصفراء والفينوفايبرات، وذلك لأنها مُرخصةٌ للاستعمال في الأطفال، ولمخاوفِ تتعلق بسلامة استعمال الأدويةِ الأخرى،[55] وعلى الرغم من هذا، فإنَّ الستاتين يبدو آمنًا وفعالًا،[56][57] ويُمكن استعمالهُ في الأطفال الأكبرِ سنًا كما في البالغين.[9][55]
في بدايةِ 2006، أوصت لجنةٌ من الخبراءِ بالعلاجِ المركب مبكرًا بفصادة البروتين الدهني منخفض الكثافة والستاتين ومثبطاتِ امتصاص الكوليسترول في الأطفال ذوي الخطرِ الأعلى من فرط كوليسترول الدم العائلي متماثل الزيجوت.[58]
الانتشار
حسبَ علم الوبائيات، فإنَّ الانتشار العالمي لفرط كوليسترول الدم العائلي حوالي 10 ملايين شخص.[14] حسبُ مُعظم الدراساتِ السُكانية، فإنَّ فرط كوليسترول الدم العائلي مُتغاير الزيجوت يحدثُ بحوالي 1:500 شَخص، وليسَ شرطًا تتطورُ جميعِ الأعراض،[8] أما النوعُ متماثل الزيجوت، فإنهُ يحدث بحوالي 1:1,000,000 شخص.[8][9]
يزدادُ شيوعُ طفراتِ مستقبل البروتين الدهني منخفض الكثافة في مجموعاتٍ سُكانية مُعينة، ويُفترضُ أنَّ هذا الأمر بسببِ ظاهرة جينية تُعرف باسم تأثير المؤسس، والتي تأسست من قبلِ مجموعةٍ صغيرة من الأفراد، واحدٌ منهم أو أكثر حاملٌ للطفرة. يمتلكُ الأفريقان والكنديون الفرنسيون واللبنانيون والفنلنديون والمسيحيون معدلاتٍ عالية من الطفراتِ الخاصة والتي تجعلُ فرط كوليسترول الدم العائلي شائعًا بينهم، أما طفراتُ صميم البروتين الدُهني بي فهيَ شائعةٌ في أوروبا الوسطى.[8]
التاريخ
يُعتبر الطبيب النَرويجي كارل مولر أولَ من ربطَ بين العلاماتِ الجسمية وارتفاعِ مستويات الكوليسترول والوراثة الصبغية الجسدية السائدة، وكانَ ذلك عام 1938.[59] في أوائلِ سبعيناتِ وثمانيناتِ القرن العشرين، وصفَ الطبيبان جوزف غولدشتاين ومايكل براون السبب الجيني لفرط كوليسترول الدم العائلي، حيثُ وجدا في البداية زيادةً في نشاطِ مختزلة 3-هيدروكسي-3-ميثيل جلوتاريل تميم الإنزيم أ (HMG-CoA reductase)، إلا أنَّ الدراساتِ أظهرت أنَّ هذا لم يُفسر مستوياتِ الكوليسترول الشاذة جدًا لدى المُصابين بفرط كوليسترول الدم العائلي،[60] ثُمَ تحولَ التركيزُ إلى دراسةِ ارتباطِ البروتين الدهني مُنخفض الكثافة معَ مُستقبله، وآثارِ ضعفِ الارتباط على الأيض، وثبتَ أنَّ هذا الأمر يتعلق بالآلية الكامنة وراء فرط كوليسترول الدم العائلي.[61] فيما بعد، حُددت العديد من الطفراتِ في البروتين عن طريقِ تسلسله،[18] وفي عام 1985، حصلا على جائزة نوبل في الطب لاكتشافهما مستقبل البروتين الدهني منخفض الكثافة وتأثيره على أيض البروتينات الدُهنية.[62]


ملاحظات
- ^ أو فَرْطُ الكُولِسْتِرُولمِيَّة العائليّ[3][63] أو فَرْطُ الكوليسترول بالدمِ العائِليّ[3] أو فَرْطُ كوليستيرول الدَّم العائِلِيّ[1][2][3] أو فَرْطُ كولِسترول الدَّم العائِلِيّ.[64]
- ^ يسمى البروتين الدُهني منخفض الكثافة الكوليسترول السيء؛ لأنه في حالِ وجودِ مستوياتٍ عالية منه، فإنها تؤدي لتراكمُ الكوليسترول في الشرايين مُسببةً تصلبًا عصيدي،[65][66] أما البروتين الدُهني مرتفع الكثافة فيُسمى الكوليسترول الجيد؛ لأنهُ يحملُ الكوليسترول من أجزاء الجسم المُختلفة نحو الكبد، ثم يقومُ الكبد بإزالة الكوليسترول من الجسم.[65][66]
- ^ الوَهدة هي الحُفرة أو المنطقة المُنخفضة أو القاع بين القمتين. يُقال مكانٌ وَهْد: أي مُنخض وبهِ هُوَّة. جمعُها: وَهَدات أو وَهْدات أو وِهاد أو وَهْد.[67]
المراجع
- "ترجمةُ (Familial hypercholesterolemia) في المعجم الطبي الموحد". مكتبة لبنان ناشرون. مؤرشف من الأصل في 19 يونيو 2018. اطلع عليه بتاريخ 18 يونيو 2018. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "ترجمةُ (Familial hypercholesterolemia) في قاموس المعاني". قاموس المعاني. مؤرشف من الأصل في 19 يونيو 2018. اطلع عليه بتاريخ 18 يونيو 2018. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "ترجمةُ (Familial hypercholesterolemia) على موقع القاموس". www.alqamoos.org. مؤرشف من الأصل في 19 يونيو 2018. اطلع عليه بتاريخ 18 يونيو 2018. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Classifications of Hyperlipidemia". مؤرشف من الأصل في 26 يونيو 2018. اطلع عليه بتاريخ 25 يونيو 2018. الوسيط
|CitationClass=تم تجاهله (مساعدة) -
"Familial Dyslipidemia". www.gbhealthwatch.com. مؤرشف من الأصل في 8 مايو 2016. اطلع عليه بتاريخ 25 يونيو 2018. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Fredrickson DS, Lees RS. A system for phenotyping hyperlipoproteinemia. Circulation 1965;31:321-327.
- Goldberg, AC; Hopkins, PN; Toth, PP; Ballantyne, CM; Rader, DJ; Robinson, JG; Daniels, SR; Gidding, SS; de Ferranti, SD; Ito, MK; McGowan, MP; Moriarty, PM; Cromwell, WC; Ross, JL; Ziajka, PE; National Lipid Association Expert Panel on Familial, Hypercholesterolemia. (يونيو 2011). "Familial hypercholesterolemia: screening, diagnosis and management of pediatric and adult patients: clinical guidance from the National Lipid Association Expert Panel on Familial Hypercholesterolemia". Journal of clinical lipidology. 5 (3 Suppl): S1-8. doi:10.1016/j.jacl.2011.04.003. PMID 21600525. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Rader DJ, Cohen J, Hobbs HH (2003). "Monogenic hypercholesterolemia: new insights in pathogenesis and treatment". J. Clin. Invest. 111 (12): 1795–803. doi:10.1172/JCI18925. PMC 161432. PMID 12813012. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Durrington P (2003). "Dyslipidaemia". Lancet. 362 (9385): 717–31. doi:10.1016/S0140-6736(03)14234-1. PMID 12957096. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Frew, J. W.; Murrell, D. F.; Haber, R. M. (2015). "Fifty shades of yellow: A review of the xanthodermatoses". International Journal of Dermatology. 54 (10): 1109–23. doi:10.1111/ijd.12945. PMID 26227781. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Tsouli SG, Kiortsis DN, Argyropoulou MI, Mikhailidis DP, Elisaf MS (2005). "Pathogenesis, detection and treatment of Achilles tendon xanthomas". Eur. J. Clin. Invest. 35 (4): 236–44. doi:10.1111/j.1365-2362.2005.01484.x. PMID 15816992. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Jansen AC, van Aalst-Cohen ES, Tanck MW, et al. (2004). "The contribution of classical risk factors to cardiovascular disease in familial hypercholesterolaemia: data in 2400 patients". J. Intern. Med. 256 (6): 482–90. doi:10.1111/j.1365-2796.2004.01405.x. PMID 15554949. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Williams RR, Hunt SC, Schumacher MC, et al. (1993). "Diagnosing heterozygous familial hypercholesterolemia using new practical criteria validated by molecular genetics". Am J Cardiol. 2 (72): 171–76. doi:10.1016/0002-9149(93)90155-6. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Repas TB, Tanner JR (February 2014). "Preventing early cardiovascular death in patients with familial hypercholesterolemia". J Am Osteopath Assoc. 114 (2): 99–108. doi:10.7556/jaoa.2014.023. PMID 24481802. مؤرشف من الأصل في 11 مارس 2014. الوسيط
|CitationClass=تم تجاهله (مساعدة) - van Aalst-Cohen ES, Jansen AC, Tanck MW, et al. (2006). "Diagnosing familial hypercholesterolaemia: the relevance of genetic testing". Eur. Heart J. 27 (18): 2240–6. doi:10.1093/eurheartj/ehl113. PMID 16825289. مؤرشف من الأصل في 4 يونيو 2009. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Genome sequencing elucidates Sardinian genetic architecture and augments association analyses for lipid and blood inflammatory markers". Nature Genetics. 47 (11): 1272–1281. 14 سبتمبر 2015. doi:10.1038/ng.3368. مؤرشف من الأصل في 30 أبريل 2019. اطلع عليه بتاريخ 23 يونيو 2018. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Moghadasian MH, Salen G, Frohlich JJ, Scudamore CH (أبريل 2002). "Cerebrotendinous xanthomatosis: a rare disease with diverse manifestations". Arch. Neurol. 59 (4): 527–9. doi:10.1001/archneur.59.4.527. PMID 11939886. مؤرشف من الأصل في 13 فبراير 2012. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Hobbs HH, Brown MS, Goldstein JL (1992). "Molecular genetics of the LDLR gene in familial hypercholesterolemia". Hum. Mutat. 1 (6): 445–66. doi:10.1002/humu.1380010602. PMID 1301956. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Vega GL, Grundy SM (1986). "In vivo evidence for reduced binding of low density lipoproteins to receptors as a cause of primary moderate hypercholesterolemia". J. Clin. Invest. 78 (5): 1410–4. doi:10.1172/JCI112729. PMC 423848. PMID 3771801. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Abifadel M, Varret M, Rabès JP, et al. (2003). "Mutations in PCSK9 cause autosomal dominant hypercholesterolemia". Nat. Genet. 34 (2): 154–6. doi:10.1038/ng1161. PMID 12730697. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Seidah NG, Khatib AM, Prat A (2006). "The proprotein convertases and their implication in sterol and/or lipid metabolism". Biol. Chem. 387 (7): 871–7. doi:10.1515/BC.2006.110. PMID 16913836. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Khachadurian AK, Uthman SM (1973). "Experiences with the homozygous cases of familial hypercholesterolemia. A report of 52 patients". Nutr Metab. 15 (1): 132–40. doi:10.1159/000175431. PMID 4351242. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Brown MS, Goldstein JL (1974). "Familial hypercholesterolemia: defective binding of lipoproteins to cultured fibroblasts associated with impaired regulation of 3-hydroxy-3-methylglutaryl coenzyme A reductase activity". Proc. Natl. Acad. Sci. U.S.A. 71 (3): 788–92. doi:10.1073/pnas.71.3.788. PMC 388099. PMID 4362634. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Scientific Steering Committee on behalf of the Simon Broome Register Group (1991). "Risk of fatal coronary heart disease in familial hypercholesterolaemia". BMJ. 303 (6807): 893–6. doi:10.1136/bmj.303.6807.893. PMC 1671226. PMID 1933004. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Sijbrands EJ, Westendorp RG, Defesche JC, de Meier PH, Smelt AH, Kastelein JJ (2001). "Mortality over two centuries in large pedigree with familial hypercholesterolaemia: family tree mortality study". BMJ. 322 (7293): 1019–23. doi:10.1136/bmj.322.7293.1019. PMC 31037. PMID 11325764. مؤرشف من الأصل في 30 أبريل 2010. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Jansen AC, van Aalst-Cohen ES, Tanck MW, et al. (2005). "Genetic determinants of cardiovascular disease risk in familial hypercholesterolemia". Arterioscler. Thromb. Vasc. Biol. 25 (7): 1475–81. doi:10.1161/01.ATV.0000168909.44877.a7. PMID 15879303. مؤرشف من الأصل في 27 مايو 2011. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Wiklund, O.; Angelin, B.; Olofsson, S. O.; Eriksson, M.; Fager, G.; Berglund, L.; Bondjers, G. (يونيو 1990). "Apolipoprotein(a) and ischaemic heart disease in familial hypercholesterolaemia". Lancet. 335 (8702): 1360–1363. doi:10.1016/0140-6736(90)91242-3. PMID 1971660. مؤرشف من الأصل (Free full text) في 24 يونيو 2018. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Seed, M.; Hoppichler, F.; Reaveley, D.; Mccarthy, S.; Thompson, G. R.; Boerwinkle, E.; Utermann, G. (مايو 1990). "Relation of serum lipoprotein(a) concentration and apolipoprotein(a) phenotype to coronary heart disease in patients with familial hypercholesterolemia". The New England Journal of Medicine. 322 (21): 1494–1499. doi:10.1056/NEJM199005243222104. ISSN 0028-4793. PMID 2139920. مؤرشف من الأصل (Free full text) في 5 مارس 2016. الوسيط
|CitationClass=تم تجاهله (مساعدة) - O'Malley JP, Maslen CL, Illingworth DR (19 مايو 1998). "Angiotensin-converting enzyme DD genotype and cardiovascular disease in heterozygous familial hypercholesterolemia". Circulation. 97 (18): 1780–3. doi:10.1161/01.CIR.97.18.1780. PMID 9603531. مؤرشف من الأصل في 8 يونيو 2011. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Besseling, J; Sjouke, B; Kastelein, JJ (أغسطس 2015). "Screening and treatment of familial hypercholesterolemia - Lessons from the past and opportunities for the future (based on the Anitschkow Lecture 2014)". Atherosclerosis. 241 (2): 597–606. doi:10.1016/j.atherosclerosis.2015.06.011. PMID 26115072. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Marks D, Wonderling D, Thorogood M, Lambert H, Humphries SE, Neil HA (يونيو 2002). "Cost effectiveness analysis of different approaches of screening for familial hypercholesterolaemia". BMJ. 324 (7349): 1303. doi:10.1136/bmj.324.7349.1303. PMC 113765. PMID 12039822. مؤرشف من الأصل في 15 سبتمبر 2009. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Umans-Eckenhausen MA, Defesche JC, Sijbrands EJ, Scheerder RL, Kastelein JJ (يناير 2001). "Review of first 5 years of screening for familial hypercholesterolaemia in the Netherlands". Lancet. 357 (9251): 165–8. doi:10.1016/S0140-6736(00)03587-X. PMID 11213091. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Marks D, Thorogood M, Neil HA, Wonderling D, Humphries SE (مارس 2003). "Comparing costs and benefits over a 10 year period of strategies for familial hypercholesterolaemia screening". J Public Health Med. 25 (1): 47–52. doi:10.1093/pubmed/fdg010. PMID 12669918. مؤرشف من الأصل (PDF) في 6 فبراير 2009. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Wald, David S.; Bestwick, Jonathan P.; Wald, Nicholas J. (2007-09-20). "Child-parent screening for familial hypercholesterolaemia: screening strategy based on a meta-analysis". BMJ (باللغة الإنجليزية). 335 (7620): 599. doi:10.1136/bmj.39300.616076.55. ISSN 0959-8138. PMC 1989026. PMID 17855284. مؤرشف من الأصل في 24 يونيو 2018. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Saenger, Amy K. (2012-08-01). "Universal Lipid Screening in Children and Adolescents: A Baby Step toward Primordial Prevention?". Clinical Chemistry (باللغة الإنجليزية). 58 (8): 1179–1181. doi:10.1373/clinchem.2012.182287. ISSN 0009-9147. PMID 22510399. مؤرشف من الأصل في 11 مايو 2020. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Caroline Parkinson (27 أكتوبر 2016). "Toddlers 'should get heart risk test'". BBC News. مؤرشف من الأصل في 25 سبتمبر 2018. اطلع عليه بتاريخ 27 أكتوبر 2016. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Wald, David S.; Bestwick, Jonathan P.; Morris, Joan K.; Whyte, Ken; Jenkins, Lucy; Wald, Nicholas J. (2016). "Child–Parent Familial Hypercholesterolemia Screening in Primary Care". New England Journal of Medicine. 375 (17): 1628–1637. doi:10.1056/NEJMoa1602777. ISSN 0028-4793. PMID 27783906. الوسيط
|CitationClass=تم تجاهله (مساعدة)
- المعهد الوطني للصحة وتفوق الرعاية. Clinical guideline 71: Familial hypercholesterolaemia. London, 2008.
- Kastelein JJ, Akdim F, Stroes ES, et al. (أبريل 2008). "Simvastatin with or without ezetimibe in familial hypercholesterolemia". N. Engl. J. Med. 358 (14): 1431–43. doi:10.1056/NEJMoa0800742. PMID 18376000. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Scientific Steering Committee on behalf of the Simon Broome Register Group (1999). "Mortality in treated heterozygous familial hypercholesterolaemia: implications for clinical management". Atherosclerosis. 142 (1): 105–12. doi:10.1016/S0021-9150(98)00200-7. PMID 9920511. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Neil A, Cooper J, Betteridge J, et al. (نوفمبر 2008). "Reductions in all-cause, cancer, and coronary mortality in statin-treated patients with heterozygous familial hypercholesterolaemia: a prospective registry study". Eur. Heart J. 29 (21): 2625–33. doi:10.1093/eurheartj/ehn422. PMC 2577142. PMID 18840879. مؤرشف من الأصل في 20 ديسمبر 2016. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Ito, MK; Santos, RD (16 مايو 2016). "PCSK9 inhibition with monoclonal antibodies-modern management of hypercholesterolemia". Journal of clinical pharmacology. Online first. doi:10.1002/jcph.766. PMC 5215586. PMID 27195910. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Marais AD, Blom DJ, Firth JC (يناير 2002). "Statins in homozygous familial hypercholesterolemia". Curr Atheroscler Rep. 4 (1): 19–25. doi:10.1007/s11883-002-0058-7. PMID 11772418. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Bilheimer DW, Goldstein JL, Grundy SM, Starzl TE, Brown MS (ديسمبر 1984). "Liver transplantation to provide low-density-lipoprotein receptors and lower plasma cholesterol in a child with homozygous familial hypercholesterolemia". N. Engl. J. Med. 311 (26): 1658–64. doi:10.1056/NEJM198412273112603. PMC 2975980. PMID 6390206. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Revell SP, Noble-Jamieson G, Johnston P, Rasmussen A, Jamieson N, Barnes ND (نوفمبر 1995). "Liver transplantation for homozygous familial hypercholesterolaemia". Arch. Dis. Child. 73 (5): 456–8. doi:10.1136/adc.73.5.456. PMC 1511367. PMID 8554367. الوسيط
|CitationClass=تم تجاهله (مساعدة) - López-Santamaria M, Migliazza L, Gamez M, et al. (أبريل 2000). "Liver transplantation in patients with homozygotic familial hypercholesterolemia previously treated by end-to-side portocaval shunt and ileal bypass". J. Pediatr. Surg. 35 (4): 630–3. doi:10.1053/jpsu.2000.0350630. PMID 10770402. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Buchwald H, Varco RL, Boen JR, et al. (يونيو 1998). "Effective lipid modification by partial ileal bypass reduced long-term coronary heart disease mortality and morbidity: five-year posttrial follow-up report from the POSCH. Program on the Surgical Control of the Hyperlipidemias". Arch. Intern. Med. 158 (11): 1253–61. doi:10.1001/archinte.158.11.1253. PMID 9625405. مؤرشف من الأصل في 8 فبراير 2012. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Bilheimer DW, Goldstein JL, Grundy SM, Brown MS (ديسمبر 1975). "Reduction in cholesterol and low density lipoprotein synthesis after portacaval shunt surgery in a patient with homozygous familial hypercholesterolemia". J. Clin. Invest. 56 (6): 1420–30. doi:10.1172/JCI108223. PMC 333120. PMID 172531. مؤرشف من الأصل في 24 يونيو 2018. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Cuchel M, Bloedon LT, Szapary PO, et al. (يناير 2007). "Inhibition of microsomal triglyceride transfer protein in familial hypercholesterolemia". N. Engl. J. Med. 356 (2): 148–56. doi:10.1056/NEJMoa061189. PMID 17215532. مؤرشف من الأصل في 27 فبراير 2009. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "FDA approves new orphan drug for rare cholesterol disorder". 31 ديسمبر 2012. مؤرشف من الأصل في 11 مايو 2020. اطلع عليه بتاريخ 24 يونيو 2018. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Pollack, Andrew (29 January 2013) F.D.A. Approves Genetic Drug to Treat Rare Disease The New York Times, Retrieved 31 January 2013 نسخة محفوظة 24 يونيو 2018 على موقع واي باك مشين.
- Staff (29 January 2013) FDA approves new orphan drug Kynamro to treat inherited cholesterol disorder U.S. Food and Drug Administration, Retrieved 31 January 2013 [وصلة مكسورة] نسخة محفوظة 06 مارس 2017 على موقع واي باك مشين.
- Grossman M, Rader DJ, Muller DW, et al. (نوفمبر 1995). "A pilot study of ex vivo gene therapy for homozygous familial hypercholesterolaemia". Nat. Med. 1 (11): 1148–54. doi:10.1038/nm1195-1148. PMID 7584986. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Mabuchi H, Koizumi J, Shimizu M, Takeda R (فبراير 1989). "Development of coronary heart disease in familial hypercholesterolemia". Circulation. 79 (2): 225–32. doi:10.1161/01.CIR.79.2.225. PMID 2914343. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Greene O, Durrington P (مايو 2004). "Clinical management of children and young adults with heterozygous familial hypercholesterolaemia in the UK". J R Soc Med. 97 (5): 226–9. doi:10.1258/jrsm.97.5.226. PMC 1079462. PMID 15121812. مؤرشف من الأصل في 05 فبراير 2009. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Rodenburg J, Vissers MN, Wiegman A, Trip MD, Bakker HD, Kastelein JJ (أغسطس 2004). "Familial hypercholesterolemia in children". Curr. Opin. Lipidol. 15 (4): 405–11. doi:10.1097/01.mol.0000137228.92396.f3. PMID 15243213. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Wiegman A, Hutten BA, de Groot E, et al. (يوليو 2004). "Efficacy and safety of statin therapy in children with familial hypercholesterolemia: a randomized controlled trial". JAMA. 292 (3): 331–7. doi:10.1001/jama.292.3.331. PMID 15265847. مؤرشف من الأصل في 2 نوفمبر 2010. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Kavey RE, Allada V, Daniels SR, et al. (ديسمبر 2006). "Cardiovascular risk reduction in high-risk pediatric patients: a scientific statement from the American Heart Association Expert Panel on Population and Prevention Science; the Councils on Cardiovascular Disease in the Young, Epidemiology and Prevention, Nutrition, Physical Activity and Metabolism, High Blood Pressure Research, Cardiovascular Nursing, and the Kidney in Heart Disease; and the Interdisciplinary Working Group on Quality of Care and Outcomes Research: endorsed by the American Academy of Pediatrics". Circulation. 114 (24): 2710–38. doi:10.1161/CIRCULATIONAHA.106.179568. PMID 17130340. مؤرشف من الأصل في 8 يونيو 2011. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Müller C (1938). "Xanthoma, hypercholesterolemia, angina pectoris". Acta Med Scandinav. 95 Suppl (89): 75–84. doi:10.1111/j.0954-6820.1938.tb19279.x. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Goldstein JL, Brown MS (أكتوبر 1973). "Familial hypercholesterolemia: identification of a defect in the regulation of 3-hydroxy-3-methylglutaryl coenzyme A reductase activity associated with overproduction of cholesterol". Proc. Natl. Acad. Sci. U.S.A. 70 (10): 2804–8. doi:10.1073/pnas.70.10.2804. PMC 427113. PMID 4355366. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Brown MS, Goldstein JL (يناير 1976). "Receptor-mediated control of cholesterol metabolism". Science. 191 (4223): 150–4. doi:10.1126/science.174194. PMID 174194. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Nobelprize.org. "Medicine 1985". مؤرشف من الأصل في 9 فبراير 2018. اطلع عليه بتاريخ 28 فبراير 2008. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "ترجمةُ (Familial hypercholesterolemia) في معجم مرعشي الطبي الكبير". مكتبة لبنان ناشرون. مؤرشف من الأصل في 19 يونيو 2018. اطلع عليه بتاريخ 18 يونيو 2018. الوسيط
|CitationClass=تم تجاهله (مساعدة) - يُوسف حِتّي; أحمَد شفيق الخَطيب (2008). قامُوس حِتّي الطِبي للجَيب. بيروت، لبنان: مكتبة لبنان ناشرون. صفحة 198. ISBN 995310235X. الوسيط
|CitationClass=تم تجاهله (مساعدة);|access-date=بحاجة لـ|url=(مساعدة) - "LDL: The "Bad" Cholesterol". MedlinePlus. مؤرشف من الأصل في 29 أغسطس 2019. اطلع عليه بتاريخ 19 يونيو 2018. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "HDL (Good), LDL (Bad) Cholesterol and Triglycerides". www.heart.org. مؤرشف من الأصل في 28 يوليو 2018. اطلع عليه بتاريخ 19 يونيو 2018. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "تعريف و معنى كلمة وهدة باللغة العربية في معجم المعاني الجامع والمعجم الوسيط واللغة العربية المعاصر". قاموس المعاني. مؤرشف من الأصل في 22 يونيو 2018. اطلع عليه بتاريخ 22 يونيو 2018. الوسيط
|CitationClass=تم تجاهله (مساعدة)
وصلات خارجية
- فرط كوليسترول الدم العائلي - تقرير مشاورة منظمة الصحة العالمية - حول تشخيص وعلاج فرط كوليسترول الدم العائلي (1998).
- قاعدة بيانات لجميع طفراتِ LDLR المعروفة (يحتفظُ بها المركز الطبي لجامعة لايدن، وباستضافة كلية لندن الجامعية).
 بوابة طب
بوابة طب