تقنيات النسخ
تقنيات النسخ: هي التقنيات المستخدمة لدراسة نسخة الكائن الحي، وهي مجموعة نسخ الحمض النووي الريبي (RNA) الخاصة به. يتم تسجيل محتوى المعلومات للكائن الحي في الحمض النووي لجينومه ويتم التعبير عنه من خلال النسخ. هنا يعمل (mRNA) كجزيء وسيط عابر في شبكة المعلومات بينما تؤدي (RNAs) الغير مشفرة وظائف متنوعة إضافية. تعمل التقنية على أخذ لقطة من النص في وقت إجمالي النصوص الموجودة في الخلية.
توفر تقنيات النسخ على نطاق واسع العمليات الخلوية النشطة والخاملة. يكمن التحدي الرئيسي في علم الأحياء الجزيئي في فهم كيف يمكن أن يؤدي نفس الجينوم إلى ظهور أنواع مختلفة من الخلايا وكيف يتم تنظيم التعبير الجيني. بدأت المحاولات الأولى لدراسة النصوص الكاملة في بداية التسعينيات، حيث أدت التطورات التكنولوجية اللاحقة منذ أواخر التسعينيات إلى تغيير المجال بشكل مستمر، وجعلت مجال تقنيات النسخ تخصصًا واسع الانتشار في العلوم البيولوجية. هناك نوعان من التقنيات المعاصرة الرئيسية في هذا المجال: المصفوفات الدقيقة ، وهي التي تحدد مجموعة من المتواليات المحددة مسبقًا، و (RNA-Seq) ، التي تستخدم التسلسل عالي الإنتاجية لتسجيل جميع النصوص. مع تطور التكنولوجيا، زاد حجم البيانات التي تنتجها كل تجربة نسخ. نتيجة لذلك، تم تكييف طرق تحليل البيانات بشكل طردي لتحليل كميات كبيرة بشكل متزايد من البيانات حيث تكون أكثر دقة وكفاءة. نمت قواعد بيانات النسخ وزادت فائدتها مع جمع المزيد من النسخ النصية ومشاركتها من قبل الباحثين. سيكون من المستحيل تقريبًا تفسير المعلومات الواردة في النسخة النصية بدون السيرعلى سياق التجارب السابقة.
إن قياس التعبير عن جينات الكائن الحي في الأنسجة أو الظروف والأوقات المختلفة، يعطي معلومات حول كيفية تنظيم الجينات ويكشف عن تفاصيل بيولوجيا الكائن الحي. يمكن استخدامه أيضًا لاستنتاج وظائف الجينات التي لم يتم توضيحها مسبقًا. مكّن تحليل تقنيات النسخ من دراسة كيفية تغير التعبير الجيني في الكائنات الحية المختلفة وكان له دور فعال في فهم الأمراض التي تصيب الإنسان. يسمح تحليل التعبير الجيني في مجمله باكتشاف اتجاهات منسقة واسعة لا يمكن تمييزها بمقايسات أكثر استهدافًا.
تاريخ تقنيات النسخ
تميز علم النسخ أنه أعاد صياغة ما هو ممكن في كل عقد وزمن، وجعلت التقنيات السابقة عفا عليها الزمن. نُشرت المحاولة الأولى لالتقاط نسخة جزئية من نسخة بشرية في عام 1991 وأبلغت عن وجود (609) تسلسل من (mRNA) من الدماغ البشري.[2] في عام 2008 ، تم نشر نسختين بشريتين، مؤلفة من ملايين التسلسلات المشتقة من النسخ والتي تغطي (16000) جينًا، وبحلول عام 2015 تم نشر النسخ لمئات الأفراد. يتم الآن بشكل روتيني إنشاء نسخ من حالات المرض المختلفة أو الأنسجة أو حتى الخلايا المفردة. هذا التطور الكبير في علم النسخ ما كان إلا نتاج تطور التكنولوجيا الحديثة بشكل دقيق ونمو الاقتصاد بشكل ملحوظ وكبير. ما قبل تقنيات النسخ:
تم إجراء دراسات على النصوص الفردية قبل عدة سنوات من ظهورمنهج تقنيات النسخ. تم جمع مكتبات نصوص (silkmoth mRNA) وتحويلها إلى حمض نووي تكميلي (cDNA) للتخزين باستخدام النسخ العكسي في أواخر السبعينيات. أما في الثمانينيات، تم استخدام التسلسل منخفض الإنتاجية باستخدام طريقة (Sanger) لتسلسل النصوص العشوائية، وإنتاج علامات تسلسل معبر عنها بصيغة (ESTs) (expressed sequence tags). كانت طريقة (Sanger) للتسلسل سائدة حتى ظهور طرق عالية الإنتاجية مثل التسلسل بالتوليف (Solexa / Illumina). برزت (ESTs) خلال التسعينيات كطريقة فعالة لتحديد المحتوى الجيني للكائن الحي دون تسلسل الجينوم بأكمله. تم قياس كميات النصوص الفردية باستخدام النشاف الشمالي، ومصفوفات غشاء النايلون، وأساليب النسخ العكسي الكمي (PCR ،RT-qPCR) لاحقًا، ولكن هذه الطرق شاقة ويمكنها فقط التقاط قسم فرعي صغير من النسخة. وبالتالي، فإن الطريقة التي يتم بها التعبير عن النص ككل وتنظيمه ظلت غير معروفة حتى تم تطوير تقنيات إنتاجية أعلى وأكثر فعالية.
التجارب الحديثة
تم استخدام كلمة "ترانسكريبتوم" التي تعني تقنيات التسخ لأول مرة في التسعينيات. في عام 1995 ، تم تطوير واحدة من أقدم طرق النسخ المستندة إلى التسلسل، وهي التحليل التسلسلي للتعبير الجيني (SAGE) (serial analysis of gene expression)، والتي عملت من خلال تسلسل سانجر (Sanger sequencing) لشظايا النسخ العشوائية المتسلسلة. تم تحديد النصوص من خلال مطابقة الأجزاء مع الجينات المعروفة. تم أيضًا استخدام متغير من (SAGE) باستخدام تقنيات التسلسل عالية التطور والإنتاجية يسمى بالتحليل التعبير الجيني الرقمي ، لفترة وجيزة. ومع ذلك، فقد تم تجاوز هذه الأساليب إلى حد كبير من خلال التسلسل عالي الإنتاجية للنصوص الكاملة، والتي قدمت معلومات إضافية حول بنية النص مثل متغيرات لصق وغيره.[3]
تطور التقنيات المعاصرة
| RNA- تسلسل | المصفوفات الدقيقة (microarray) | |
|---|---|---|
| الإنتاجية | من يوم واحد إلى أسبوع واحد لكل تجربة | يوم إلى يومين لكل تجربة |
| كمية إدخال RNA | منخفض ~ 1 نانوغرام إجمالي الحمض النووي الريبي [7] | عالية ~ 1 ميكروغرام مرنا [8] |
| كثافة اليد العاملة | مرتفع (تحضير العينات وتحليل البيانات) | منخفض |
| علم مسبق | لا شيء مطلوب، على الرغم من أن تسلسل الجينوم / النسخ المرجعي مفيد | مطلوب جينوم مرجعي / نسخة لتصميم المجسات |
| دقة الكميات | ~ 90٪ (مقيدة بالتغطية التسلسلية) [9] | > 90٪ (مقيدة بدقة الكشف عن التألق) |
| دقة التسلسل | يمكن لـ RNA-Seq اكتشاف تعدد الأشكال ومتغيرات لصق (محدودة بدقة تسلسل تصل إلى 99٪ تقريبًا) | يمكن للمصفوفات المتخصصة اكتشاف متغيرات لصق الرنا المرسال (مقيدة بتصميم المجس والتهجين المتبادل) |
| حساسية | نص واحد لكل مليون (تقريبي، محدود بتغطية التسلسل) | نص واحد لكل ألف (تقريبي، محدود باكتشاف التألق) |
| مدى ديناميكي | 100000: 1 (محدود بتغطية التسلسل) [10] | 1000: 1 (محدود بالتشبع الفلوري) |
| استنساخ التقنية | > 99٪ [11][12] | > 99٪ [13][14] |
تم تطوير التقنيات المعاصرة السائدة، المصفوفات الدقيقة (microarrays) و تسلسل RNA ، في منتصف التسعينيات والعقد الأول من القرن الحادي والعشرين. تم نشر المصفوفات الدقيقة التي تقيس وفرة مجموعة محددة من النصوص من خلال تهجينها إلى مجموعة من المجسات التكميلية لأول مرة في عام (1995). سمحت تقنية المصفوفات الدقيقة بفحص آلاف النسخ في وقت واحد وبتكلفة مخفضة بشكل كبير لكل جين وتوفير الأيدي العاملة. كانت كل من مصفوفات أليغنوكليوتيد المرقطة (spotted oligonucleotide arrays) ومصفوفات أفيميتريكس (Affymetrix) عالية الكثافة هي الطريقة الفضلى للتنميط النسخي حتى أواخر العقد الأول من القرن الحادي والعشرين. خلال هذه الفترة، تم إنتاج مجموعة من المصفوفات الدقيقة لتغطية الجينات المعروفة في الكائنات الحية النموذجية أو ذات الأهمية الاقتصادية. أدى التقدم في تصميم وتصنيع المصفوفات إلى تحسين خصوصية المجسات والسماح باختبار المزيد من الجينات على مجموعة واحدة. أدى التقدم في الكشف عن التألق إلى زيادة دقة وحساسية القياس النصوص ذات الوفرة المنخفضة.
يتم إنشاء تسلسل RNA عن طريق النسخ العكسي لـ كلًا من RNA و وتسلسل (cDNAs) الناتجة في المختبر[6] يتم اشتقاق وفرة النسخ الناتجة من عدد التهم من كل نص. لذلك فقد تأثرت هذه التقنية بشكل كبير بتطوير تقنيات التسلسل عالية الإنتاجية .[15][16] كان تسلسل التوقيع المتوازي (MPSS) مثالًا مبكرًا يعتمد على توليد من 16-20 متواليات bp عبر سلسلة معقدة من التهجين، [17] [note 1] واستخدمت في عام (2004) للتحقق من صحة التعبير عن عشرة آلاف جين في (Arabidopsis thaliana) .[18] تم نشر أقدم عمل لتسلسل RNA في عام 2006 مع مائة ألف نسخة متسلسلة باستخدام تقنية 454 .[19] كانت هذه تغطية كافية لتحديد وفرة النسخ النسبية. بدأت شعبية RNA-Seq في الزيادة بعد عام 2008 عندما سمحت تقنيات Solexa / Illumina الجديدة بتسجيل مليار تسلسل نسخ.[20][21][22] يسمح هذا العائد الآن بتحديد ومقارنة النسخ البشرية.[23]
جمع البيانات
يمكن توليد البيانات عن نصوص الحمض النووي الريبي (mRNA) من خلال واحد من مبدأين مهمين: تسلسل النصوص الفردية ( ESTs ، أو RNA-Seq) أو تهجين النصوص إلى مجموعة مرتبة من تحقيقات النيوكليوتيدات (المصفوفات الدقيقة) (microarray).[4]
عزل الحمض النووي الريبي mRNA
تتطلب جميع طرق النسخ أن يتم عزل RNA أولاً عن الكائن التجريبي قبل تسجيل النصوص. على الرغم من أن الأنظمة البيولوجية متنوعة بشكل لا يصدق، فإن تقنيات استخراج الحمض النووي الريبي متشابهة إلى حد كبير وتتضمن التعطيل الميكانيكي للخلايا أو الأنسجة، وتعطيل RNase بأملاح متقلبة، [24] اضطراب الجزيئات الكبيرة والمجمعات النوكليوتيدية المعقدة، وفصل الحمض النووي الريبي عن الجزيئات الحيوية غير المرغوب فيها بما في ذلك الحمض النووي DNA، والتركيز من الحمض النووي الريبي عن طريق الترسيب من المحلول أو الشطف من مصفوفة صلبة .[25] يمكن أيضًا معالجة الحمض النووي الريبي المعزول باستخدام الدناز لهضم أي آثار للحمض النووي.[26] من الضروري إثراء الحمض النووي الريبي المرسال لأن مستخلصات الحمض النووي الريبي الكلية عادة ما تكون 98٪ من الحمض النووي الريبوزي .[27] يمكن إجراء الإثراء للنصوص بواسطة طرق تقارب poly-A أو عن طريق استنفاد RNA الريبوسومي باستخدام تحقيقات خاصة بالتسلسل.[28] قد يؤثر الحمض النووي الريبي المتدهور أو الشائب على نتائج المصب ؛ على سبيل المثال، سيؤدي تخصيب mRNA من العينات المتدهورة إلى استنفاد 5 'نهايات mRNA وإشارة غير متساوية عبر طول النص. يعد التجميد المفاجئ للأنسجة قبل عزل الحمض النووي الريبي أمرًا نموذجيًا، ويتم الحرص على تقليل التعرض لإنزيمات RNase بمجرد اكتمال العزل.
علامات التسلسل المعبر عنها
علامة التسلسل المعبر عنها (EST) هي تسلسل نيوكليوتيد قصير يتم إنشاؤه من نسخة واحدة من RNA. يتم نسخ الحمض النووي الريبي (RNA) أولاً على أنه DNA تكميلي (cDNA) بواسطة إنزيم النسخ العكسي قبل تسلسل cDNA الناتج.[29] نظرًا لأنه يمكن جمع ESTs دون معرفة مسبقة بالكائن الحي الذي أتت منه، فيمكن صنعها من خليط من الكائنات الحية أو العينات البيئية.[30] على الرغم من استخدام طرق الإنتاجية العالية الآن، إلا أن مكتبات EST تقدم معلومات تسلسل لتصميمات المصفوفات الدقيقة المبكرة ؛ على سبيل المثال، تم تصميم ميكروأري الشعير من 350.000 EST متسلسلة مسبقًا.[31]
التحليل التسلسلي والغطاء للتعبير الجيني (SAGE / CAGE)
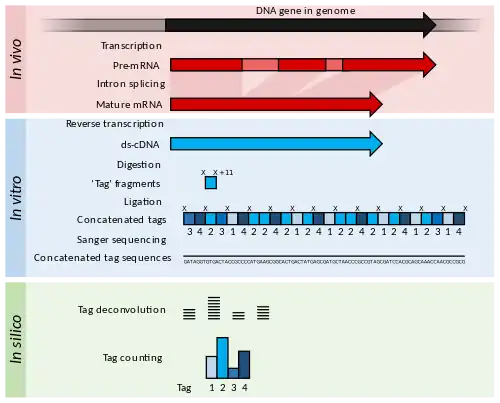
كان التحليل( التسلسلي للتعبير الجيني (SAGE) تطويرًا لمنهجية EST لزيادة إنتاجية العلامات التي تم إنشاؤها والسماح بدخول بعض التقدير الكمي لوفرة النص.[33] cDNA [33] يتم توليدها من الحمض النووي الريبي mRNA ولكن بعد ذلك يتم هضمها إلى 11 pp tag باستخدام إنزيمات التقييد التي تقطع الحمض النووي في تسلسل معين، و 11 زوج من القاعدة على طول هذا التسلسل. بعد ذلك يتم ضم cDNA في سلاسل (> 500 bp) ومتسلسلة باستخدام طرق منخفضة، ولكن قراءة طويلة المدى مثل تسلسل Sanger . ثم يتم تقسيم التسلسلات مرة أخرى إلى علامات 11 نقطة أساس باستخدام برامج الكمبيوتر في عملية تسمى deconvolution . إذا كان الجينوم المرجعي متاحًا، فقد تتطابق هذه العلامات مع الجين المقابل في الجينوم. إذا كان الجينوم المرجعي غير متاح، فيمكن استخدام العلامات مباشرة كعلامات تشخيصية إذا وجد أنه يتم التعبير عنها بشكل مختلف في حالة المرض.
مصطلح التعبير الجيني لتحليل الغطاء (CAGE) هي أحد أشكال SAGE التي تسلسل العلامات من نهاية 5 ' من نسخة mRNA فقط.[34] لذلك، يمكن تحديد موقع بدء نسخ الجينات عند محاذاة العلامات إلى جينوم مرجعي. يعد تحديد مواقع بدء الجينات مفيدًا لتحليل المروج واستنساخ cDNAs كاملة الطول.
تنتج طرق كلًا من SAGE و CAGE معلومات عن جينات أكثر مما كان ممكنًا في السابق عند تسلسل ESTs الفردية، ولكن تحضير العينات وتحليل البيانات عادة ما يكونان أكثر كثافة في العمل.[34]
المصفوفات الدقيقة
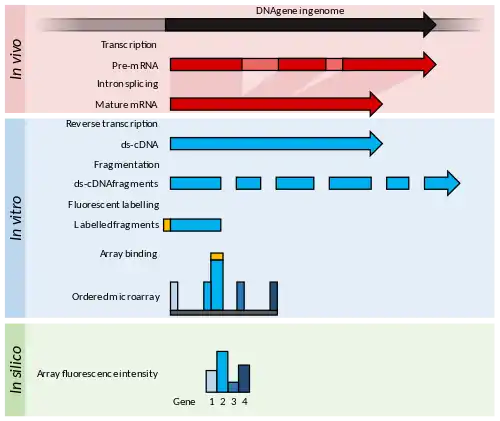
المبادئ والتطورات
تتكون المصفوفات الدقيقة من أوليغومرات نيوكليوتيد قصيرة، تُعرف باسم " المجسات " ، والتي يتم ترتيبها عادةً في شبكة على شريحة زجاجية.[35] يتم تحديد وفرة نسخة عن طريق تهجين النصوص المسمى فلوريسنتلي لهذه التحقيقات.[36] تشير شدة التألق في كل موقع مسبار على الصفيف إلى وفرة النسخ لتسلسل التحقيق هذا.
تتطلب المصفوفات الدقيقة بعض المعرفة الجينومية من الكائن الحي محل الاهتمام، على سبيل المثال، في شكل تسلسل الجينوم المشروح، أو مكتبة من ESTs التي يمكن استخدامها لإنشاء تحقيقات للمصفوفة.[37]
طرق
عادةً ما تندرج المصفوفات الدقيقة الخاصة بالنسخ في إحدى فئتين عريضتين: المصفوفات المرقطة منخفضة الكثافة أو المصفوفات القصيرة عالية الكثافة. يتم الاستدلال على وفرة النسخ من شدة التألق المستمدة من النصوص ذات العلامات الفلورية التي ترتبط بالمصفوفة.[37]
تتميز المصفوفات ذات الكثافة المنخفضة المرقطة عادةً بقطرات بيكوليتر [note 2] لمجموعة من cDNAs المنقى والمصفوفة على سطح شريحة زجاجية.[38] هذه المجسات أطول من تلك الخاصة بالمصفوفات عالية الكثافة ولا يمكنها تحديد أحداث الربط البديلة . تستخدم المصفوفات المرقطة نوعين مختلفين من الفلوروفور لتسمية عينات الاختبار والتحكم، وتستخدم نسبة التألق لحساب مقياس نسبي للوفرة.[39] تستخدم المصفوفات عالية الكثافة تسمية فلورية واحدة، ويتم تهجين كل عينة واكتشافها على حدة.[40] وقد شاع صفائف عالية الكثافة من قبل Affymetrix GeneChip مجموعة، حيث يتم كميا كل محضر من قبل العديد من القصير 25 -mer تحقيقات أن معا مقايسة جين واحد.[41]
كانت صفائف NimbleGen عبارة عن مصفوفة عالية الكثافة تم إنتاجها بواسطة طريقة الكيمياء الضوئية بدون قناع، والتي سمحت بتصنيع مرن للمصفوفات بأعداد صغيرة أو كبيرة. كانت هذه المصفوفات تحتوي على 100000s من 45 إلى 85 مير وتم تهجينها بعينة ذات لون واحد لتحليل التعبير.[42] تضمنت بعض التصميمات ما يصل إلى 12 مصفوفة مستقلة لكل شريحة.
تسلسل RNA
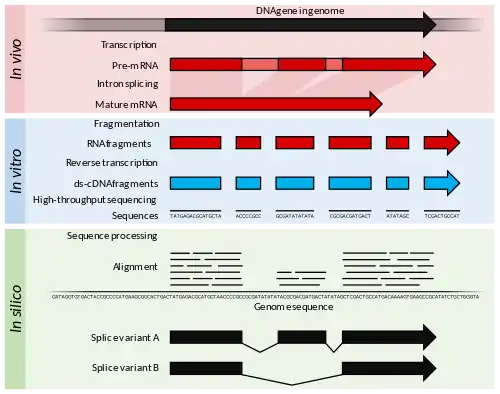
المبادئ والتطورات
يشير RNA-Seq إلى مزيج من منهجية التسلسل عالية الإنتاجية مع الأساليب الحسابية لالتقاط وقياس النصوص الموجودة في مستخلص الحمض النووي الريبي.[6] يبلغ طول تسلسل النوكليوتيدات الناتج حوالي 100 نقطة أساس، ولكن يمكن أن يتراوح من 30 نقطة أساس إلى أكثر من 10000 نقطة أساس اعتمادًا على طريقة التسلسل المستخدمة. يستفيد RNA-Seq من أخذ عينات عميقة للنسخة مع العديد من الأجزاء القصيرة من نسخة للسماح بإعادة البناء الحسابي لنسخة RNA الأصلية عن طريق محاذاة القراءات مع جينوم مرجعي أو مع بعضها البعض ( تجميع de novo ).[15] يمكن قياس كمية الرنا منخفضة الوفرة وعالية الوفرة في تجربة RNA-Seq ( نطاق ديناميكي من 5 أوامر من حيث الحجم ) - وهي ميزة رئيسية على نسخ المصفوفات الدقيقة. بالإضافة إلى ذلك، فإن كميات إدخال الحمض النووي الريبي (RNA) أقل بكثير بالنسبة لـ RNA-Seq (كمية نانوجرام) مقارنة بالمصفوفات الدقيقة (كمية ميكروغرام) ، مما يسمح بفحص أدق للهياكل الخلوية وصولاً إلى مستوى الخلية المفردة عند دمجها مع التضخيم الخطي لـ cDNA.[7][43] من الناحية النظرية، لا يوجد حد أعلى للقياس الكمي في RNA-Seq ، وضوضاء الخلفية منخفضة جدًا لقراءات 100 نقطة أساس في المناطق غير المتكررة.
يمكن استخدام RNA-Seq لتحديد الجينات داخل الجينوم ، أو تحديد الجينات النشطة في نقطة زمنية معينة، ويمكن استخدام أعداد القراءة لنمذجة مستوى التعبير الجيني النسبي بدقة. لقد تحسنت منهجية RNA-Seq باستمرار، في المقام الأول من خلال تطوير تقنيات تسلسل الحمض النووي لزيادة الإنتاجية والدقة وطول القراءة.[44] منذ الأوصاف الأولى في عامي 2006 و 2008 ، [19][45] تم اعتماد RNA-Seq بسرعة وتجاوز المصفوفات الدقيقة باعتبارها تقنية النسخ السائدة في عام 2015.[46]
أدى البحث عن بيانات النسخ على مستوى الخلايا الفردية إلى حدوث تقدم في أساليب إعداد مكتبة RNA-Seq ، مما أدى إلى تطورات مثيرة في الحساسية. يتم الآن وصف نسخ الخلية المفردة بشكل جيد وقد تم تمديدها إلى RNA-Seq في الموقع حيث يتم استجواب نسخ الخلايا الفردية مباشرة في الأنسجة الثابتة .[47]
طرق
تم إنشاء RNA-Seq بالتنسيق مع التطور السريع لمجموعة من تقنيات تسلسل الحمض النووي عالية الإنتاجية.[48] ومع ذلك، قبل تسلسل نسخ RNA المستخرجة، يتم تنفيذ عدة خطوات معالجة رئيسية. تختلف الطرق في استخدام إثراء النص، والتجزئة، والتضخيم، والتسلسل الفردي أو المزدوج، وما إذا كان يجب الحفاظ على معلومات الخيوط.
يمكن زيادة حساسية تجربة RNA-Seq عن طريق إثراء فئات من الحمض النووي الريبي (RNA) ذات الأهمية واستنفاد الحمض النووي الريبي المعروف الوفير. يمكن فصل جزيئات mRNA باستخدام تحقيقات oligonucleotides التي تربط ذيول poly-A الخاصة بها . بدلا من ذلك، RIBO استنفاد يمكن استخدامها لإزالة تحديدا وفيرة ولكن غير المفيدة الرنا الريباسي (rRNAs) من خلال التهجين لتحقيقات مصممة خصيصا ل الأصنوفة في تسلسل الريباسي محددة (على سبيل المثال الثدييات الريباسي، مصنع الريباسي). ومع ذلك، يمكن أن يؤدي استنفاد الضلع أيضًا إلى بعض التحيز عبر الاستنفاد غير المحدد للنصوص غير المستهدفة.[49] يمكن تنقية RNAs الصغيرة، مثل micro RNAs ، بناءً على حجمها عن طريق الرحلان الكهربائي للهلام والاستخراج.
نظرًا لأن mRNAs أطول من أطوال القراءة الخاصة بأساليب التسلسل عالية الإنتاجية النموذجية، يتم عادةً تجزئة النصوص قبل التسلسل.[50] تعد طريقة التجزئة جانبًا رئيسيًا في بناء مكتبة التسلسل. يمكن تحقيق التجزئة عن طريق التحليل المائي الكيميائي ، أو الإرذاذ، أو الصوتنة، أو النسخ العكسي باستخدام نيوكليوتيدات متسلسلة . بدلاً من ذلك، يمكن إجراء التجزئة وعلامات (كدنا) في وقت واحد باستخدام إنزيمات ترانسبوزيز .[51]
أثناء التحضير للتسلسل، يمكن تضخيم نسخ (كدنا) من النصوص بواسطة تفاعل البوليميراز المتسلسل لإثراء الأجزاء التي تحتوي على تسلسل المحول المتوقع 5 'و 3'.[52] يستخدم التضخيم أيضًا للسماح بتسلسل كميات إدخال منخفضة جدًا من RNA ، وصولاً إلى أقل من 50 بيكوغرام في التطبيقات المتطرفة.[53] يمكن استخدام ضوابط الزيادة في RNAs المعروفة لتقييم مراقبة الجودة للتحقق من إعداد المكتبة وتسلسلها، من حيث محتوى GC ، وطول الجزء، بالإضافة إلى التحيز بسبب موضع التجزئة داخل النص.[54] المعرّفات الجزيئية الفريدة (UMIs) عبارة عن تسلسلات عشوائية قصيرة تُستخدم لتمييز أجزاء التسلسل بشكل فردي أثناء إعداد المكتبة بحيث يكون كل جزء مميز فريدًا.[55] توفر UMIs مقياسًا مطلقًا للتقدير الكمي، وفرصة لتصحيح تحيز التضخيم اللاحق الذي تم تقديمه أثناء إنشاء المكتبة، وتقدير حجم العينة الأولي بدقة. تعتبر UMIs مناسبة بشكل خاص لنسخة RNA-Seq أحادية الخلية، حيث يتم تقييد كمية إدخال الحمض النووي الريبي (RNA) والتضخيم الموسع للعينة مطلوبًا.[56][57][58]
بمجرد أن يتم تحضير جزيئات النص، يمكن تسلسلها في اتجاه واحد فقط (طرف واحد) أو كلا الاتجاهين (نهاية مزدوجة). عادةً ما يكون إنتاج التسلسل أحادي النهاية أسرع، وأرخص من التسلسل ثنائي النهاية وكافٍ لتقدير مستويات التعبير الجيني. ينتج عن التسلسل المزدوج النهاية محاذاة / تجميعات أكثر قوة، وهو أمر مفيد للتعليق التوضيحي للجينات واكتشاف الشكل الإسوي للنسخ.[6] تحافظ أساليب RNA-Seq الخاصة بالجدار على معلومات حبلا لنسخة متسلسلة.[59] بدون معلومات الخيط، يمكن محاذاة القراءات مع موضع الجين ولكن لا تخبر في أي اتجاه يتم نسخ الجين. يعد Stranded-RNA-Seq مفيدًا لفك تشفير النسخ للجينات التي تتداخل في اتجاهات مختلفة ولإجراء تنبؤات جينية أكثر قوة في الكائنات غير النموذجية.
| منصة | الافراج التجاري | طول القراءة النموذجي | أقصى سرعة لكل شوط | دقة قراءة واحدة | عمليات تشغيل RNA-Seq المودعة في NCBI SRA (أكتوبر 2016) [62] |
|---|---|---|---|---|---|
| 454 علوم الحياة | 2005 | 700 بي بي | 0.7 جيجابت | 99.9٪ | 3548 |
| إلومينا | 2006 | 50 - 300 سنة مضت | 900 جيجابت | 99.9٪ | 362903 |
| صلب | 2008 | 50 زوجا | 320 جيجابت | 99.9٪ | 7032 |
| ايون تورنت | 2010 | 400 نقطة أساس | 30 جيجابت | 98٪ | 1953 |
| باكبيو | 2011 | 10000 نقطة أساس | 2 جيجابت | 87٪ | 160 |
يعتمد RNA-Seq حاليًا على نسخ جزيئات RNA إلى جزيئات cDNA قبل التسلسل ؛ لذلك، الأنظمة الأساسية اللاحقة هي نفسها بالنسبة للبيانات الجينية والنسخية. وبالتالي، كان تطوير تقنيات تسلسل الحمض النووي سمة مميزة لـ RNA-Seq.[61][63][64] التسلسل المباشر للحمض النووي الريبي باستخدام التسلسل النانوي يمثل أحدث تقنيات RNA-Seq الحالية.[65][66] يمكن للتسلسل النانوي للحمض النووي الريبي أن يكتشف القواعد المعدلة التي يمكن إخفاؤها بطريقة أخرى عند تسلسل cDNA وأيضًا يلغي خطوات التضخيم التي يمكن أن تؤدي إلى التحيز.[16][67]
تعتمد حساسية ودقة تجربة RNA-Seq على عدد القراءات التي تم الحصول عليها من كل عينة.[68][69] هناك حاجة إلى عدد كبير من القراءات لضمان التغطية الكافية للنسخة، مما يتيح الكشف عن النصوص منخفضة الوفرة. يزداد التصميم التجريبي تعقيدًا عن طريق تقنيات التسلسل ذات نطاق الإخراج المحدود، والكفاءة المتغيرة لإنشاء التسلسل، وجودة التسلسل المتغير. يضاف إلى هذه الاعتبارات أن كل نوع لديه عدد مختلف من الجينات وبالتالي يتطلب إنتاج تسلسل مخصص لنسخة فعالة. حددت الدراسات المبكرة عتبات مناسبة تجريبيًا، ولكن مع نضوج التكنولوجيا، تم التنبؤ بالتغطية المناسبة حسابياً عن طريق تشبع النسخ. الطريقة الأكثر فاعلية لتحسين الكشف عن التعبير التفاضلي في الجينات منخفضة التعبير هي إضافة المزيد من التكرارات البيولوجية بدلاً من إضافة المزيد من القراءات.[70] المعايير الحالية التي أوصى بها مشروع Encyclopedia of DNA Elements (ENCODE) هي لتغطية الإكسوم 70 ضعفًا لمعيار RNA-Seq وتغطية exome تصل إلى 500 ضعف لاكتشاف النصوص والأشكال الإسوية النادرة.[71][72][73]
تحليل البيانات
طرق النسخ متوازية للغاية وتتطلب حسابًا كبيرًا لإنتاج بيانات ذات مغزى لكل من تجارب ميكروأري و RNA-Seq.[74][75][76][77] يتم تسجيل بيانات Microarray كصور عالية الدقة ، مما يتطلب اكتشاف الميزات والتحليل الطيفي.[78] يبلغ حجم كل من ملفات الصور الأولية Microarray حوالي 750 ميجابايت، بينما يبلغ حجم الشدة المعالجة حوالي 60 ميجابايت. يمكن أن تكشف التحقيقات القصيرة المتعددة التي تتطابق مع نسخة واحدة عن تفاصيل حول بنية intron - exon ، مما يتطلب نماذج إحصائية لتحديد مصداقية الإشارة الناتجة. تنتج دراسات RNA-Seq المليارات من سلاسل الحمض النووي القصيرة، والتي يجب أن تتماشى مع الجينومات المرجعية المكونة من ملايين إلى مليارات أزواج القواعد. يتطلب تجميع القراءات <i id="mwApQ">De novo</i> داخل مجموعة البيانات إنشاء رسوم بيانية تسلسلية معقدة للغاية.[79] عمليات RNA-Seq متكررة للغاية وتستفيد من الحساب المتوازي ولكن الخوارزميات الحديثة تعني أن أجهزة الحوسبة الاستهلاكية كافية لتجارب النسخ البسيطة التي لا تتطلب تجميعًا جديدًا للقراءات .[80] يمكن التقاط نسخة بشرية بدقة باستخدام RNA-Seq مع 30 مليون تسلسل 100 نقطة أساس لكل عينة.[68][69] قد يتطلب هذا المثال ما يقرب من 1.8 جيجا بايت من مساحة القرص لكل عينة عند تخزينها بتنسيق fastq مضغوط. ستكون بيانات العد المعالج لكل جين أصغر بكثير، أي ما يعادل شدة ميكروأري المعالجة. قد يتم تخزين بيانات التسلسل في مستودعات عامة، مثل أرشيف قراءة التسلسل (SRA).[81] يمكن تحميل مجموعات بيانات RNA-Seq عبر Gene Expression Omnibus.[82]
معالجة الصورة

يجب أن تحدد معالجة الصور بالميكروأري بشكل صحيح الشبكة العادية للميزات داخل صورة وأن تحدد بشكل مستقل شدة التألق لكل ميزة. بالإضافة إلى ذلك، يجب تحديد القطع الأثرية للصورة وإزالتها من التحليل العام. تشير شدة الإسفار مباشرة إلى وفرة كل تسلسل، حيث أن تسلسل كل مسبار في الصفيف معروف بالفعل.[84]
تتضمن الخطوات الأولى لـ RNA-seq أيضًا معالجة صور مماثلة ؛ ومع ذلك، عادةً ما يتم التعامل مع تحويل الصور إلى بيانات متسلسلة تلقائيًا بواسطة برنامج الجهاز. ينتج عن طريقة تسلسل Illumina بالتخليق مجموعة من المجموعات الموزعة على سطح خلية التدفق.[85] يتم تصوير خلية التدفق حتى أربع مرات خلال كل دورة تسلسل، مع عشرات إلى مئات الدورات في المجموع. تتشابه مجموعات خلايا التدفق مع بقع ميكروأري ويجب تحديدها بشكل صحيح خلال المراحل المبكرة من عملية التسلسل. في طريقة Roche للتسلسل الحراري ، تحدد شدة الضوء المنبعث عدد النيوكليوتيدات المتتالية في تكرار البوليمر المتجانس. هناك العديد من المتغيرات في هذه الطرق ، ولكل منها ملف تعريف خطأ مختلف للبيانات الناتجة.[86]
تحليل بيانات تسلسل RNA
تولد تجارب RNA-Seq حجمًا كبيرًا من قراءات التسلسل الأولي التي يجب معالجتها للحصول على معلومات مفيدة. يتطلب تحليل البيانات عادةً مجموعة من أدوات برمجيات المعلوماتية الحيوية (انظر أيضًا قائمة أدوات المعلوماتية الحيوية RNA-Seq ) التي تختلف وفقًا للتصميم والأهداف التجريبية. يمكن تقسيم العملية إلى أربع مراحل: مراقبة الجودة والمحاذاة والتقدير والتعبير التفاضلي.[87] يتم تشغيل معظم برامج RNA-Seq الشائعة من واجهة سطر الأوامر ، إما في بيئة Unix أو ضمن البيئة الإحصائية R / Bioconductor .[76]
رقابة الجودة
قراءات التسلسل ليست مثالية ، لذلك يجب تقدير دقة كل قاعدة في التسلسل لتحليلات المصب. يتم فحص البيانات الأولية للتأكد من: درجات الجودة للمكالمات الأساسية عالية ، ومحتوى GC يطابق التوزيع المتوقع ، والزخارف المتسلسلة القصيرة ( k-mers ) ليست ممثلة بشكل زائد ، ومعدل تكرار القراءة منخفض بشكل مقبول.[69] توجد عدة خيارات برمجية لتحليل جودة التسلسل ، بما في ذلك FastQC و FaQCs.[88][89] يمكن إزالة التشوهات (التشذيب) أو تمييزها بمعاملة خاصة أثناء العمليات اللاحقة.
توضيح
من أجل ربط تسلسل قراءة الوفرة بالتعبير عن جين معين ، يتم محاذاة تسلسل النص إلى جينوم مرجعي أو <i id="mwAtc">دي نوفو</i> محاذي لبعضها البعض إذا لم يتوفر مرجع.[90][91] تتضمن التحديات الرئيسية لبرمجيات المحاذاة سرعة كافية للسماح بمحاذاة مليارات من التسلسلات القصيرة في إطار زمني ذي مغزى ، والمرونة في التعرف على التضفير الداخلي للـ mRNA والتعامل معه ، والتخصيص الصحيح للقراءات التي تعينها على مواقع متعددة. عالجت التطورات البرمجية هذه المشكلات بشكل كبير ، كما أن الزيادات في طول قراءة التسلسل تقلل من فرصة محاذاة القراءة الغامضة. يحتفظ EBI بقائمة بمحاذاة التسلسل عالية الإنتاجية المتوفرة حاليًا.[92][93]
تتطلب محاذاة تسلسلات mRNA للنسخة الأولية المشتقة من حقيقيات النوى إلى جينوم مرجعي معالجة متخصصة لتسلسلات intron ، والتي لا توجد في mRNA الناضج.[94] تقوم أدوات محاذاة القراءة القصيرة بإجراء جولة إضافية من المحاذاة المصممة خصيصًا لتحديد الوصلات الملصقة ، ويتم إعلامها من خلال تسلسل موقع لصق الكنسي ومعلومات موقع لصق إنترون المعروفة. إن تحديد تقاطعات لصق intron يمنع القراءات من أن تكون غير محاذاة عبر تقاطعات الوصلات أو يتم التخلص منها بشكل خاطئ ، مما يسمح بمحاذاة المزيد من القراءات مع الجينوم المرجعي وتحسين دقة تقديرات التعبير الجيني. نظرًا لأن تنظيم الجينات قد يحدث على مستوى الشكل الإسوي للـ mRNA ، فإن المحاذاة الواعية للوصلة تسمح أيضًا بالكشف عن تغييرات وفرة الشكل الإسوي التي قد تُفقد في تحليل مجمّع.[95]
يمكن استخدام تجميع De novo لمحاذاة القراءات مع بعضها البعض لإنشاء تسلسلات نصية كاملة الطول دون استخدام جينوم مرجعي.[96] تشمل التحديات الخاصة بتجميع de novo متطلبات حسابية أكبر مقارنة بنسخة مرجعية ، والتحقق الإضافي من المتغيرات الجينية أو الأجزاء ، والشرح الإضافي للنصوص المجمعة. لقد ثبت أن المقاييس الأولى المستخدمة لوصف تجميعات النسخ ، مثل N50 ، مضللة [97] وأن طرق التقييم المحسنة متاحة الآن.[98][99] مقاييس تعتمد على الشرح وتقييم أفضل من اكتمال التجميع، مثل contig متبادلة أفضل ضرب العد. بمجرد تجميع de novo ، يمكن استخدام التجميع كمرجع لطرق محاذاة التسلسل اللاحقة وتحليل التعبير الجيني الكمي.
| البرمجيات | صدر | آخر تحديث | الكفاءة الحسابية | نقاط القوة والضعف |
|---|---|---|---|---|
| الواحات المخملية [100][101] | 2008 | 2011 | متطلبات ذاكرة الوصول العشوائي منخفضة ، أحادية الخيوط ، عالية | مُجمّع القراءة القصيرة الأصلي. تم استبداله الآن إلى حد كبير. |
| سوابدينوفو ترانس [91] | 2011 | 2014 | متطلبات ذاكرة الوصول العشوائي المعتدلة ومتعددة الخيوط والمتوسطة | مثال مبكر لمجمع قراءة قصيرة. تم تحديثه لتجميع النسخ. |
| Trans-ABySS [102] | 2010 | 2016 | متطلبات ذاكرة الوصول العشوائي المعتدلة ومتعددة الخيوط والمتوسطة | مناسبة للقراءات القصيرة ، ويمكنها التعامل مع النسخ المعقدة ، ويتوفر إصدار MPI المتوازي لحوسبة المجموعات. |
| الثالوث [79][103] | 2011 | 2017 | متطلبات ذاكرة الوصول العشوائي المعتدلة ومتعددة الخيوط والمتوسطة | مناسبة للقراءات القصيرة. يمكنه التعامل مع النسخ المعقدة ولكنها تتطلب ذاكرة مكثفة. |
| ميريست [104] | 1999 | 2016 | متطلبات ذاكرة الوصول العشوائي المعتدلة ومتعددة الخيوط والمتوسطة | يمكن معالجة التسلسلات المتكررة ، والجمع بين تنسيقات التسلسل المختلفة ، ويتم قبول مجموعة واسعة من أنظمة التسلسل. |
| نيوبلير [105] | 2004 | 2012 | متطلبات ذاكرة الوصول العشوائي منخفضة ، أحادية الخيط ، عالية | متخصص لاستيعاب أخطاء تسلسل البوليمر المتماثل النموذجي لمسلسلات Roche 454. |
| منضدة عمل علم الجينوم CLC [106] | 2008 | 2014 | متطلبات ذاكرة الوصول العشوائي عالية ومتعددة الخيوط ومنخفضة | لديه واجهة مستخدم رسومية ، ويمكنه الجمع بين تقنيات التسلسل المتنوعة ، ولا يحتوي على ميزات خاصة بالنسخة ، ويجب شراء ترخيص قبل الاستخدام. |
| SPAdes [107] | 2012 | 2017 | متطلبات ذاكرة الوصول العشوائي عالية ومتعددة الخيوط ومنخفضة | تستخدم لتجارب النسخ على الخلايا المفردة. |
| RSEM [108] | 2011 | 2017 | متطلبات ذاكرة الوصول العشوائي عالية ومتعددة الخيوط ومنخفضة | يمكن تقدير تكرار النصوص المقسمة بشكل بديل. سهل الاستخدام. |
| StringTie [80][109] | 2015 | 2019 | متطلبات ذاكرة الوصول العشوائي عالية ومتعددة الخيوط ومنخفضة | يمكن استخدام مزيج من طرق التجميع الموجه بالمرجع و de novo لتحديد النصوص. |
RAM - ذاكرة الوصول العشوائي ؛ MPI - واجهة تمرير الرسائل ؛ EST - علامة التسلسل المعبر عنها.
تحديد الكميات
يمكن إجراء التحديد الكمي لمحاذاة التسلسل على مستوى الجين أو إكسون أو نسخة.[70] تتضمن المخرجات النموذجية جدولًا بأعداد القراءة لكل ميزة يتم توفيرها للبرنامج ؛ على سبيل المثال ، للجينات في ملف تنسيق ميزة عامة . يمكن حساب عدد قراءة الجينات والإكسون بسهولة تامة باستخدام HTSeq ، على سبيل المثال.[111] يعتبر التحديد الكمي على مستوى النص أكثر تعقيدًا ويتطلب طرقًا احتمالية لتقدير وفرة النص الإسوي من معلومات القراءة القصيرة ؛ على سبيل المثال ، باستخدام برنامج أزرار الكم.[95] يجب تحديد القراءات التي تتماشى جيدًا مع مواقع متعددة وإما إزالتها أو محاذاتها مع أحد المواقع المحتملة أو مواءمتها مع الموقع الأكثر احتمالًا.
يمكن لبعض طرق القياس الكمي الالتفاف على الحاجة إلى محاذاة دقيقة للقراءة إلى تسلسل مرجعي تمامًا. تجمع طريقة برنامج kallisto بين المحاذاة الزائفة والقياس الكمي في خطوة واحدة تعمل على تنفيذ 2 أوامر من حيث الحجم أسرع من الأساليب المعاصرة مثل تلك المستخدمة بواسطة برنامج tophat / cufflinks ، مع عبء حسابي أقل.[112]
بمجرد توفر التعداد الكمي لكل نص ، يتم قياس التعبير الجيني التفاضلي عن طريق تطبيع البيانات والنمذجة والتحليل الإحصائي لها.[90] ستقرأ معظم الأدوات جدولًا للجينات وتقرأ الأعداد كمدخلات لها ، لكن بعض البرامج ، مثل cuffdiff ، ستقبل تنسيق خريطة المحاذاة الثنائية كمدخلات. النواتج النهائية لهذه التحليلات هي قوائم الجينات المصحوبة باختبارات زوجية مرتبطة بالتعبير التفاضلي بين العلاجات وتقديرات احتمالية هذه الاختلافات.[113]
| البرمجيات | بيئة | تخصص |
|---|---|---|
| Cuffdiff2 [90] | يونكس | تحليل النسخ الذي يتتبع التضفير البديل لـ mRNA |
| EdgeR [75] | R / موصل حيوي | أي بيانات الجينوم القائمة على العد |
| DEseq2 [114] | R / موصل حيوي | أنواع بيانات مرنة ، تكرار منخفض |
| ليما / فوم [74] | R / موصل حيوي | بيانات Microarray أو RNA-Seq ، تصميم تجربة مرن |
| ثوب الكرة [115] | R / موصل حيوي | اكتشاف النصوص فعالة وحساسة ومرنة. |
mRNA - messenger RNA.
التحقق من الصحة
يمكن التحقق من صحة تحليلات النسخ باستخدام تقنية مستقلة ، على سبيل المثال ، PCR الكمي (qPCR) ، والتي يمكن التعرف عليها وتقييمها إحصائيًا.[116] يتم قياس التعبير الجيني مقابل معايير محددة لكل من الجينات المعنية والجينات الضابطة . القياس بواسطة qPCR مشابه للقياس الذي حصل عليه RNA-Seq حيث يمكن حساب قيمة لتركيز المنطقة المستهدفة في عينة معينة. ومع ذلك ، فإن qPCR مقصور على amplicons أصغر من 300 نقطة أساس ، وعادةً ما تكون باتجاه الطرف 3 'من منطقة التشفير ، مع تجنب 3'UTR .[117] إذا كان التحقق من صحة الأشكال الإسوية للنسخة مطلوبًا ، فيجب أن يشير فحص محاذاة قراءة RNA-Seq إلى المكان الذي يمكن فيه وضع بادئات qPCR للتمييز الأقصى. ينتج عن قياس جينات التحكم المتعددة جنبًا إلى جنب مع الجينات ذات الأهمية مرجعًا ثابتًا ضمن سياق بيولوجي.[118] أظهر التحقق من صحة qPCR لبيانات RNA-Seq عمومًا أن طرق RNA-Seq المختلفة مترابطة بشكل كبير.[45][119][120]
يعد التحقق الوظيفي للجينات الرئيسية من الاعتبارات المهمة للتخطيط بعد النسخ. قد تكون أنماط التعبير الجيني لوحظت ترتبط وظيفيا إلى النمط الظاهري عن طريق مستقلة تدق إلى أسفل / الإنقاذ الدراسة في الكائن الحي من الفائدة.[121]
التطبيقات
التشخيص وتحديد ملامح المرض
شهدت استراتيجيات النسخ تطبيقًا واسعًا عبر مجالات متنوعة من البحوث الطبية الحيوية ، بما في ذلك تشخيص المرض والتنميط .[6][122] سمحت مناهج RNA-Seq بالتعرف على نطاق واسع لمواقع بدء النسخ ، واستخدام المروج البديل غير المكشوف ، وتعديلات الربط الجديدة. هذه العناصر التنظيمية مهمة في مرض الإنسان ، وبالتالي ، فإن تحديد مثل هذه المتغيرات أمر بالغ الأهمية لتفسير دراسات ارتباط المرض .[123] يمكن لـ RNA-Seq أيضًا تحديد تعدد أشكال النوكليوتيدات المفردة (SNPs) المرتبطة بالأمراض ، والتعبير الخاص بالأليل ، واندماج الجينات ، مما يساهم في فهم المتغيرات المسببة للمرض.[124]
الينقولات الرجعية هي عناصر قابلة للنقل والتي تتكاثر داخل جينومات حقيقية النواة من خلال عملية تتضمن النسخ العكسي . يمكن أن يوفر RNA-Seq معلومات حول نسخ الينقولات العكسية الذاتية التي قد تؤثر على نسخ الجينات المجاورة بواسطة آليات جينية مختلفة تؤدي إلى المرض.[125] وبالمثل، فإن إمكانية استخدام الحمض النووي الريبي تسلسل لفهم مرض المناعة ذات الصلة توسعا سريعا بسبب القدرة على تشريح السكان الخلايا المناعية وتسلسل الخلايا T و مستقبلات الخلايا B ذخيرة من المرضى.[126][127]
النسخ البشرية وعوامل المسببات المرضية
أصبح RNA-Seq لمسببات الأمراض البشرية طريقة ثابتة لقياس تغيرات التعبير الجيني ، وتحديد عوامل الضراوة الجديدة ، والتنبؤ بمقاومة المضادات الحيوية ، وكشف النقاب عن التفاعلات المناعية للمضيف الممرض .[128][129] الهدف الأساسي من هذه التقنية هو تطوير تدابير مكافحة العدوى المثلى والعلاج الفردي المستهدف.[127]
ركز تحليل النسخ في الغالب على المضيف أو الممرض تم تطبيق Dual RNA-Seq لتشكيل تعبير RNA في وقت واحد في كل من الممرض والمضيف طوال عملية العدوى. تمكن هذه التقنية من دراسة الاستجابة الديناميكية وشبكات تنظيم الجينات بين الأنواع في كل من شركاء التفاعل من الاتصال الأولي وحتى الغزو والثبات النهائي للعامل الممرض أو التطهير بواسطة الجهاز المناعي المضيف.[130][131]
استجابات البيئة
يسمح علم النسخ بتحديد الجينات والمسارات التي تستجيب للضغوط البيئية الحيوية وغير الحيوية وتتصدى لها . [121] تسمح الطبيعة غير المستهدفة للنصوص بتحديد شبكات النسخ الجديدة في الأنظمة المعقدة. على سبيل المثال ، حدد التحليل المقارن لمجموعة من سلالات الحمص في مراحل نمو مختلفة ملامح نسخ متميزة مرتبطة بضغوط الجفاف والملوحة ، بما في ذلك تحديد دور الأشكال الإسوية للنسخة من AP2 - EREBP .[132] التحقيق في التعبير الجيني خلال بيوفيلم تشكيل من الفطريات المسببة للمرض المبيضات البيض كشفت مجموعة المشترك المنظم للجينات حاسمة لإنشاء بيوفيلم والصيانة.[133]
يوفر التنميط النسخي أيضًا معلومات مهمة حول آليات مقاومة الأدوية . حدد تحليل أكثر من 1000 عزلة من Plasmodium falciparum ، وهو طفيلي خبيث مسؤول عن الملاريا في البشر ، [134] أن زيادة تنظيم استجابة البروتين غير المطوية والتقدم البطيء خلال المراحل المبكرة من دورة النمو اللاجنسي داخل الكريات الحمر ارتبطت بمقاومة الأرتيميسينين في العزلات من جنوب شرق آسيا .[135]
شرح وظيفة الجينات
كانت جميع تقنيات النسخ مفيدة بشكل خاص في تحديد وظائف الجينات وتحديد المسؤولين عن أنماط ظاهرية معينة. Transcriptomics من نبات الأرابيدوبسيس الطرز البيئية التي المعادن hyperaccumulate ارتباطا الجينات المسؤولة عن امتصاص المعادن والتسامح و التوازن مع النمط الظاهري.[136] تم استخدام تكامل مجموعات بيانات RNA-Seq عبر الأنسجة المختلفة لتحسين شرح وظائف الجينات في الكائنات الحية المهمة تجاريًا (مثل الخيار ) [137] أو الأنواع المهددة (مثل الكوالا ).[138]
لا تعتمد قراءة تجميع RNA-Seq على الجينوم المرجعي [103] ولذا فهي مثالية لدراسات التعبير الجيني للكائنات غير النموذجية ذات الموارد الجينومية غير الموجودة أو ضعيفة التطور. على سبيل المثال ، تم إنشاء قاعدة بيانات لـ SNPs المستخدمة في برامج تربية دوغلاس التنوب عن طريق تحليل نسخة دي نوفو في غياب الجينوم المتسلسل .[139] وبالمثل ، تم تحديد الجينات التي تعمل في تطوير أنسجة القلب والعضلات والعصبية في الكركند من خلال مقارنة الترانسكريبتومات لأنواع الأنسجة المختلفة دون استخدام تسلسل الجينوم.[140] يمكن أيضًا استخدام RNA-Seq لتحديد مناطق ترميز البروتين غير المعروفة سابقًا في الجينوم المتسلسل الحالي.
الشيخوخة القائمة على النسخ
التدخلات الوقائية المتعلقة بالشيخوخة غير ممكنة بدون قياس سرعة الشيخوخة الشخصية. الطريقة الأحدث والأكثر تعقيدًا لقياس معدل الشيخوخة هي باستخدام المؤشرات الحيوية المتغيرة لشيخوخة الإنسان التي تعتمد على استخدام الشبكات العصبية العميقة التي يمكن تدريبها على أي نوع من البيانات البيولوجية omics للتنبؤ بعمر الموضوع. لقد ثبت أن الشيخوخة محرك قوي لتغييرات النسخ.[141][142] عانت الساعات القديمة القائمة على النسخ من تباين كبير في البيانات ودقة منخفضة نسبيًا. ومع ذلك ، فإن النهج الذي يستخدم مقياسًا زمنيًا وثنائية الترانسكريبتومات لتحديد مجموعة الجينات التي تتنبأ بالعمر البيولوجي بدقة يسمح لها بالوصول إلى تقييم قريب من الحد النظري .
الحمض النووي الريبي الغير مشفر
يتم تطبيق Transcriptomics بشكل شائع على محتوى mRNA للخلية. ومع ذلك ، فإن نفس التقنيات قابلة للتطبيق بشكل متساوٍ على RNAs غير المشفرة (ncRNAs) التي لا تُترجم إلى بروتين ، ولكن بدلاً من ذلك لها وظائف مباشرة (مثل الأدوار في ترجمة البروتين ، وتكرار الحمض النووي ، وربط الحمض النووي الريبي ، وتنظيم النسخ ).[143][144][145][146] تؤثر العديد من ncRNAs على الحالات المرضية ، بما في ذلك السرطان وأمراض القلب والأوعية الدموية والأمراض العصبية.[147]
قواعد بيانات النسخ
تولد دراسات النسخ كميات كبيرة من البيانات التي لها تطبيقات محتملة تتجاوز بكثير الأهداف الأصلية للتجربة. على هذا النحو ، قد يتم إيداع البيانات الخام أو المعالجة في قواعد البيانات العامة لضمان فائدتها للمجتمع العلمي الأوسع. على سبيل المثال ، اعتبارًا من 2018 ، احتوى Gene Expression Omnibus على ملايين التجارب.[148]
| اسم | مضيف | البيانات | وصف |
|---|---|---|---|
| Omnibus التعبير الجيني [82] | NCBI | ميكروأري RNA-Seq | أول قاعدة بيانات transcriptomics لقبول البيانات من أي مصدر. قدم MIAME و MINSEQE معايير المجتمع التي تحدد الفوقية التجربة اللازمة لضمان تفسير وفعال التكرار .[149] |
| ArrayExpress [150] | ENA | ميكروأري | يستورد مجموعات البيانات من Gene Expression Omnibus ويقبل عمليات الإرسال المباشرة. يتم تخزين البيانات المعالجة والبيانات الوصفية للتجربة في ArrayExpress ، بينما يتم الاحتفاظ بقراءات التسلسل الأولي في ENA. يتوافق مع معايير MIAME و MINSEQE.[151] |
| أطلس التعبير [152] | EBI | ميكروأري RNA-Seq | قاعدة بيانات التعبير الجيني الخاصة بالأنسجة للحيوانات والنباتات. يعرض التحليلات الثانوية والتصور ، مثل الإثراء الوظيفي لمصطلحات Gene Ontology أو مجالات InterPro أو المسارات. روابط لبيانات وفرة البروتين عند توفرها. |
| جينيفستيجاتور [153] | برعاية خاصة | ميكروأري RNA-Seq | يحتوي على تنظيمات يدوية لمجموعات بيانات النسخ العامة ، مع التركيز على البيانات الطبية والبيولوجية النباتية. يتم تطبيع التجارب الفردية عبر قاعدة البيانات الكاملة للسماح بمقارنة التعبير الجيني عبر تجارب متنوعة. تتطلب الوظائف الكاملة شراء ترخيص ، مع وصول مجاني إلى وظائف محدودة. |
| المرجع [154] | DDBJ | الكل | نسخ الإنسان والفأر والجرذان من 40 عضوًا مختلفًا. يتم تصور التعبير الجيني كخرائط حرارية معروضة على تمثيلات ثلاثية الأبعاد للهياكل التشريحية. |
| NONCODE [155] | noncode.org | RNA- تسلسل | الحمض النووي الريبي غير المشفر (ncRNAs) باستثناء الحمض الريبي النووي الريبي والـ rRNA. |
NCBI - المركز الوطني لمعلومات التكنولوجيا الحيوية ؛ EBI - المعهد الأوروبي للمعلوماتية الحيوية ؛ DDBJ - بنك بيانات DNA الياباني ؛ ENA - أرشيف النوكليوتيدات الأوروبي ؛ MIAME - الحد الأدنى من المعلومات حول تجربة ميكروأري ؛ MINSEQE - الحد الأدنى من المعلومات حول تجربة SEQuencing عالية الإنتاجية للنيوكليوتيدات.
انظر أيضًا
المراجع
- "Medline trend: automated yearly statistics of PubMed results for any query". dan.corlan.net. مؤرشف من الأصل في 12 أغسطس 2020. اطلع عليه بتاريخ 05 أكتوبر 2016. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Complementary DNA sequencing: expressed sequence tags and human genome project". Science. 252 (5013): 1651–6. June 1991. Bibcode:1991Sci...252.1651A. doi:10.1126/science.2047873. PMID 2047873. S2CID 13436211. الوسيط
|CitationClass=تم تجاهله (مساعدة)صيانة CS1: عرض-المؤلفون (link) - "Transcriptomics in the RNA-seq era". Current Opinion in Chemical Biology. 17 (1): 4–11. February 2013. doi:10.1016/j.cbpa.2012.12.008. PMID 23290152. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Comparing bioinformatic gene expression profiling methods: microarray and RNA-Seq". Medical Science Monitor Basic Research. 20: 138–42. August 2014. doi:10.12659/MSMBR.892101. PMID 25149683. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Comparison of RNA-Seq and microarray in transcriptome profiling of activated T cells". PLOS ONE. 9 (1): e78644. 2014. Bibcode:2014PLoSO...978644Z. doi:10.1371/journal.pone.0078644. PMID 24454679. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "RNA-Seq: a revolutionary tool for transcriptomics". Nature Reviews Genetics. 10 (1): 57–63. January 2009. doi:10.1038/nrg2484. PMID 19015660. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "CEL-Seq: single-cell RNA-Seq by multiplexed linear amplification". Cell Reports. 2 (3): 666–73. September 2012. doi:10.1016/j.celrep.2012.08.003. PMID 22939981. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "A novel, sensitive detection system for high-density microarrays using dendrimer technology". Physiological Genomics. 3 (2): 93–9. August 2000. doi:10.1152/physiolgenomics.2000.3.2.93. PMID 11015604. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Illumina (2011-07-11). "RNA-Seq Data Comparison with Gene Expression Microarrays" (PDF). European Pharmaceutical Review. مؤرشف من الأصل (PDF) في 28 سبتمبر 2020. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Comparison of microarrays and RNA-seq for gene expression analyses of dose-response experiments". Toxicological Sciences. 137 (2): 385–403. February 2014. doi:10.1093/toxsci/kft249. PMID 24194394. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "RNA-seq: an assessment of technical reproducibility and comparison with gene expression arrays". Genome Research. 18 (9): 1509–17. September 2008. doi:10.1101/gr.079558.108. PMID 18550803. الوسيط
|CitationClass=تم تجاهله (مساعدة) - SEQC/MAQC-III Consortium (September 2014). "A comprehensive assessment of RNA-seq accuracy, reproducibility and information content by the Sequencing Quality Control Consortium". Nature Biotechnology. 32 (9): 903–14. doi:10.1038/nbt.2957. PMID 25150838. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Reproducibility of microarray data: a further analysis of microarray quality control (MAQC) data". BMC Bioinformatics. 8: 412. October 2007. doi:10.1186/1471-2105-8-412. PMID 17961233. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Independence and reproducibility across microarray platforms". Nature Methods. 2 (5): 337–44. May 2005. doi:10.1038/nmeth757. PMID 15846360. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Transcriptomics in the RNA-seq era". Current Opinion in Chemical Biology. 17 (1): 4–11. February 2013. doi:10.1016/j.cbpa.2012.12.008. PMID 23290152. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "RNA sequencing: advances, challenges and opportunities". Nature Reviews Genetics. 12 (2): 87–98. February 2011. doi:10.1038/nrg2934. PMID 21191423. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Gene expression analysis by massively parallel signature sequencing (MPSS) on microbead arrays". Nature Biotechnology. 18 (6): 630–4. June 2000. doi:10.1038/76469. PMID 10835600. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Analysis of the transcriptional complexity of Arabidopsis thaliana by massively parallel signature sequencing". Nature Biotechnology. 22 (8): 1006–11. August 2004. doi:10.1038/nbt992. PMID 15247925. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Analysis of the prostate cancer cell line LNCaP transcriptome using a sequencing-by-synthesis approach". BMC Genomics. 7: 246. September 2006. doi:10.1186/1471-2164-7-246. PMID 17010196. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "A global view of gene activity and alternative splicing by deep sequencing of the human transcriptome". Science. 321 (5891): 956–60. August 2008. Bibcode:2008Sci...321..956S. doi:10.1126/science.1160342. PMID 18599741. الوسيط
|CitationClass=تم تجاهله (مساعدة)صيانة CS1: عرض-المؤلفون (link) - "Mapping and quantifying mammalian transcriptomes by RNA-Seq". Nature Methods. 5 (7): 621–8. July 2008. doi:10.1038/nmeth.1226. PMID 18516045. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Dynamic repertoire of a eukaryotic transcriptome surveyed at single-nucleotide resolution". Nature. 453 (7199): 1239–43. June 2008. Bibcode:2008Natur.453.1239W. doi:10.1038/nature07002. PMID 18488015. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "A global view of gene activity and alternative splicing by deep sequencing of the human transcriptome". Science. 321 (5891): 956–60. August 2008. Bibcode:2008Sci...321..956S. doi:10.1126/science.1160342. PMID 18599741. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction". Analytical Biochemistry. 162 (1): 156–9. April 1987. doi:10.1016/0003-2697(87)90021-2. PMID 2440339. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "The single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction: twenty-something years on". Nature Protocols. 1 (2): 581–5. 2006. doi:10.1038/nprot.2006.83. PMID 17406285. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Use of reverse transcriptase polymerase chain reaction to monitor expression of intronless genes". BioTechniques. 9 (3): 262, 264, 266–8. September 1990. PMID 1699561. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Isolation of messenger RNA". RNA Isolation and Characterization Protocols. 86. 1998. صفحات 61–4. doi:10.1385/0-89603-494-1:61. ISBN 978-0-89603-494-5. PMID 9664454. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Comparison of RNA-Seq by poly (A) capture, ribosomal RNA depletion, and DNA microarray for expression profiling". BMC Genomics. 15: 419. June 2014. doi:10.1186/1471-2164-15-419. PMID 24888378. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Expressed sequence tags—ESTablishing bridges between genomes". Trends in Genetics. 14 (1): 4–7. January 1998. doi:10.1016/S0168-9525(97)01355-3. PMID 9448457. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Some examples of environmental samples include: sea water, soil, or air.
- "A new resource for cereal genomics: 22K barley GeneChip comes of age". Plant Physiology. 134 (3): 960–8. March 2004. doi:10.1104/pp.103.034462. PMID 15020760. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Transcriptomics technologies". PLOS Computational Biology. 13 (5): e1005457. May 2017. Bibcode:2017PLSCB..13E5457L. doi:10.1371/journal.pcbi.1005457. PMID 28545146. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Serial analysis of gene expression". Science. 270 (5235): 484–7. October 1995. Bibcode:1995Sci...270..484V. doi:10.1126/science.270.5235.484. PMID 7570003. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Cap analysis gene expression for high-throughput analysis of transcriptional starting point and identification of promoter usage". Proceedings of the National Academy of Sciences of the United States of America. 100 (26): 15776–81. December 2003. Bibcode:2003PNAS..10015776S. doi:10.1073/pnas.2136655100. PMID 14663149. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "A critical comparison of protein microarray fabrication technologies". The Analyst. 139 (6): 1303–26. March 2014. Bibcode:2014Ana...139.1303R. doi:10.1039/c3an01577g. PMID 24479125. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Bio-microarray fabrication techniques—a review". Critical Reviews in Biotechnology. 26 (4): 237–59. 2006-10-01. doi:10.1080/07388550600978358. PMID 17095434. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "DNA microarray technology: devices, systems, and applications". Annual Review of Biomedical Engineering. 4: 129–53. 2002. doi:10.1146/annurev.bioeng.4.020702.153438. PMID 12117754. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Robotic spotting of cDNA and oligonucleotide microarrays". Trends in Biotechnology. 23 (7): 374–9. July 2005. doi:10.1016/j.tibtech.2005.04.002. PMID 15978318. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "A DNA microarray system for analyzing complex DNA samples using two-color fluorescent probe hybridization". Genome Research. 6 (7): 639–45. July 1996. doi:10.1101/gr.6.7.639. PMID 8796352. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Expression monitoring by hybridization to high-density oligonucleotide arrays". Nature Biotechnology. 14 (13): 1675–80. December 1996. doi:10.1038/nbt1296-1675. PMID 9634850. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Summaries of Affymetrix GeneChip probe level data". Nucleic Acids Research. 31 (4): 15e–15. February 2003. doi:10.1093/nar/gng015. PMID 12582260. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Analysis of chromosome breakpoints in neuroblastoma at sub-kilobase resolution using fine-tiling oligonucleotide array CGH". Genes, Chromosomes & Cancer. 44 (3): 305–19. November 2005. doi:10.1002/gcc.20243. PMID 16075461. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Exponential scaling of single-cell RNA-seq in the past decade". Nature Protocols. 13 (4): 599–604. April 2018. doi:10.1038/nprot.2017.149. PMID 29494575. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Tachibana, Chris (2015-08-18). "Transcriptomics today: Microarrays, RNA-seq, and more". Science. 349 (6247): 544. Bibcode:2015Sci...349..544T. doi:10.1126/science.opms.p1500095. مؤرشف من الأصل في 30 ديسمبر 2017. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "The transcriptional landscape of the yeast genome defined by RNA sequencing". Science. 320 (5881): 1344–9. June 2008. Bibcode:2008Sci...320.1344N. doi:10.1126/science.1158441. PMID 18451266. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "An investigation of biomarkers derived from legacy microarray data for their utility in the RNA-seq era". Genome Biology. 15 (12): 523. December 2014. doi:10.1186/s13059-014-0523-y. PMID 25633159. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Highly multiplexed subcellular RNA sequencing in situ". Science. 343 (6177): 1360–3. March 2014. Bibcode:2014Sci...343.1360L. doi:10.1126/science.1250212. PMID 24578530. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Next-generation DNA sequencing". Nature Biotechnology. 26 (10): 1135–45. October 2008. doi:10.1038/nbt1486. PMID 18846087. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "IVT-seq reveals extreme bias in RNA sequencing". Genome Biology. 15 (6): R86. June 2014. doi:10.1186/gb-2014-15-6-r86. PMID 24981968. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Systematic comparison of three methods for fragmentation of long-range PCR products for next generation sequencing". PLOS ONE. 6 (11): e28240. 2011. Bibcode:2011PLoSO...628240K. doi:10.1371/journal.pone.0028240. PMID 22140562. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "ClickSeq: Fragmentation-Free Next-Generation Sequencing via Click Ligation of Adaptors to Stochastically Terminated 3'-Azido cDNAs". Journal of Molecular Biology. 427 (16): 2610–6. August 2015. doi:10.1016/j.jmb.2015.06.011. PMID 26116762. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "The impact of amplification on differential expression analyses by RNA-seq". Scientific Reports. 6: 25533. May 2016. Bibcode:2016NatSR...625533P. doi:10.1038/srep25533. PMID 27156886. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Evaluation of commercially available RNA amplification kits for RNA sequencing using very low input amounts of total RNA". Journal of Biomolecular Techniques. 26 (1): 4–18. April 2015. doi:10.7171/jbt.15-2601-001. PMID 25649271. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Synthetic spike-in standards for RNA-seq experiments". Genome Research. 21 (9): 1543–51. September 2011. doi:10.1101/gr.121095.111. PMID 21816910. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Counting absolute numbers of molecules using unique molecular identifiers". Nature Methods. 9 (1): 72–4. November 2011. doi:10.1038/nmeth.1778. PMID 22101854. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "mRNA-Seq whole-transcriptome analysis of a single cell". Nature Methods. 6 (5): 377–82. May 2009. doi:10.1038/nmeth.1315. PMID 19349980. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Quantitative single-cell RNA-seq with unique molecular identifiers". Nature Methods. 11 (2): 163–6. February 2014. doi:10.1038/nmeth.2772. PMID 24363023. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Massively parallel single-cell RNA-seq for marker-free decomposition of tissues into cell types". Science. 343 (6172): 776–9. February 2014. Bibcode:2014Sci...343..776J. doi:10.1126/science.1247651. PMID 24531970. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Comprehensive comparative analysis of strand-specific RNA sequencing methods". Nature Methods. 7 (9): 709–15. September 2010. doi:10.1038/nmeth.1491. PMID 20711195. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "A tale of three next generation sequencing platforms: comparison of Ion Torrent, Pacific Biosciences and Illumina MiSeq sequencers". BMC Genomics. 13: 341. July 2012. doi:10.1186/1471-2164-13-341. PMID 22827831. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Comparison of next-generation sequencing systems". Journal of Biomedicine & Biotechnology. 2012: 251364. 2012. doi:10.1155/2012/251364. PMID 22829749. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "SRA". مؤرشف من الأصل في 14 ديسمبر 2020. اطلع عليه بتاريخ 06 أكتوبر 2016. الوسيط
|CitationClass=تم تجاهله (مساعدة)The NCBI Sequence Read Archive (SRA) was searched using “RNA-Seq[Strategy]” and one of "LS454[Platform]”, “Illumina[platform]”, "ABI Solid[Platform]”, "Ion Torrent[Platform]”, "PacBio SMRT"[Platform]” to report the number of RNA-Seq runs deposited for each platform. - "Performance comparison of benchtop high-throughput sequencing platforms". Nature Biotechnology. 30 (5): 434–9. May 2012. doi:10.1038/nbt.2198. PMID 22522955. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Coming of age: ten years of next-generation sequencing technologies". Nature Reviews Genetics. 17 (6): 333–51. May 2016. doi:10.1038/nrg.2016.49. PMID 27184599. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Highly parallel direct RNA sequencing on an array of nanopores". Nature Methods (باللغة الإنجليزية). 15 (3): 201–206. March 2018. doi:10.1038/nmeth.4577. PMID 29334379. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "A complete bacterial genome assembled de novo using only nanopore sequencing data". Nature Methods. 12 (8): 733–5. August 2015. doi:10.1038/nmeth.3444. PMID 26076426. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Direct RNA sequencing". Nature. 461 (7265): 814–8. October 2009. Bibcode:2009Natur.461..814O. doi:10.1038/nature08390. PMID 19776739. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Calculating sample size estimates for RNA sequencing data". Journal of Computational Biology. 20 (12): 970–8. December 2013. doi:10.1089/cmb.2012.0283. PMID 23961961. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "A survey of best practices for RNA-seq data analysis". Genome Biology. 17: 13. January 2016. doi:10.1186/s13059-016-0881-8. PMID 26813401. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Comprehensive evaluation of differential gene expression analysis methods for RNA-seq data". Genome Biology. 14 (9): R95. 2013. doi:10.1186/gb-2013-14-9-r95. PMID 24020486. الوسيط
|CitationClass=تم تجاهله (مساعدة) - ENCODE Project Consortium; Aldred, Shelley F.; Collins, Patrick J.; Davis, Carrie A.; Doyle, Francis; Epstein, Charles B.; Frietze, Seth; Harrow, Jennifer; Kaul, Rajinder (September 2012). "An integrated encyclopedia of DNA elements in the human genome". Nature. 489 (7414): 57–74. Bibcode:2012Natur.489...57T. doi:10.1038/nature11247. PMID 22955616. الوسيط
|CitationClass=تم تجاهله (مساعدة)صيانة CS1: عرض-المؤلفون (link) - "ENCODE data at the ENCODE portal". Nucleic Acids Research. 44 (D1): D726–32. January 2016. doi:10.1093/nar/gkv1160. PMID 26527727. الوسيط
|CitationClass=تم تجاهله (مساعدة)صيانة CS1: عرض-المؤلفون (link) - "ENCODE: Encyclopedia of DNA Elements". encodeproject.org. مؤرشف من الأصل في 20 نوفمبر 2020. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "limma powers differential expression analyses for RNA-sequencing and microarray studies". Nucleic Acids Research. 43 (7): e47. April 2015. doi:10.1093/nar/gkv007. PMID 25605792. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "edgeR: a Bioconductor package for differential expression analysis of digital gene expression data". Bioinformatics. 26 (1): 139–40. January 2010. doi:10.1093/bioinformatics/btp616. PMID 19910308. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Orchestrating high-throughput genomic analysis with Bioconductor". Nature Methods. 12 (2): 115–21. February 2015. doi:10.1038/nmeth.3252. PMID 25633503. الوسيط
|CitationClass=تم تجاهله (مساعدة)صيانة CS1: عرض-المؤلفون (link) - Smyth, G. K. (2005). "Limma: Linear Models for Microarray Data". Bioinformatics and Computational Biology Solutions Using R and Bioconductor. (باللغة الإنجليزية). Springer, New York, NY. صفحات 397–420. doi:10.1007/0-387-29362-0_23. ISBN 9780387251462. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Steve., Russell (2008). Microarray Technology in Practice. Meadows, Lisa A. Burlington: Elsevier. ISBN 9780080919768. OCLC 437246554. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis". Nature Protocols. 8 (8): 1494–512. August 2013. doi:10.1038/nprot.2013.084. PMID 23845962. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "StringTie enables improved reconstruction of a transcriptome from RNA-seq reads". Nature Biotechnology (باللغة الإنجليزية). 33 (3): 290–5. March 2015. doi:10.1038/nbt.3122. PMID 25690850. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "The Sequence Read Archive: explosive growth of sequencing data". Nucleic Acids Research. 40 (Database issue): D54–6. January 2012. doi:10.1093/nar/gkr854. PMID 22009675. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Gene Expression Omnibus: NCBI gene expression and hybridization array data repository". Nucleic Acids Research. 30 (1): 207–10. January 2002. doi:10.1093/nar/30.1.207. PMID 11752295. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Petrov, Anton; Shams, Soheil (2004-11-01). "Microarray Image Processing and Quality Control". Journal of VLSI Signal Processing Systems for Signal, Image and Video Technology. 38 (3): 211–226. doi:10.1023/B:VLSI.0000042488.08307.ad. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Petrov, Anton; Shams, Soheil (2004). "Microarray Image Processing and Quality Control". The Journal of VLSI Signal Processing-Systems for Signal, Image, and Video Technology. 38 (3): 211–226. doi:10.1023/B:VLSI.0000042488.08307.ad. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Kwon, Young Min; Ricke, Steven (2011). High-Throughput Next Generation Sequencing. 733. SpringerLink. doi:10.1007/978-1-61779-089-8. ISBN 978-1-61779-088-1. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Sequence-specific error profile of Illumina sequencers". Nucleic Acids Research. 39 (13): e90. July 2011. doi:10.1093/nar/gkr344. PMID 21576222. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "RNA-Seq: revelation of the messengers". Trends in Plant Science. 18 (4): 175–9. April 2013. doi:10.1016/j.tplants.2013.02.001. PMID 23481128. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "FastQC: A Quality Control tool for High Throughput Sequence Data". Babraham Bioinformatics. 2010. مؤرشف من الأصل في 24 نوفمبر 2020. اطلع عليه بتاريخ 23 مايو 2017. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Rapid evaluation and quality control of next generation sequencing data with FaQCs". BMC Bioinformatics. 15: 366. November 2014. doi:10.1186/s12859-014-0366-2. PMID 25408143. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Differential analysis of gene regulation at transcript resolution with RNA-seq". Nature Biotechnology. 31 (1): 46–53. January 2013. doi:10.1038/nbt.2450. PMID 23222703. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "SOAPdenovo-Trans: de novo transcriptome assembly with short RNA-Seq reads". Bioinformatics. 30 (12): 1660–6. June 2014. arXiv:1305.6760. doi:10.1093/bioinformatics/btu077. PMID 24532719. الوسيط
|CitationClass=تم تجاهله (مساعدة) - HTS Mappers. http://www.ebi.ac.uk/~nf/hts_mappers/ نسخة محفوظة 12 نوفمبر 2020 على موقع واي باك مشين.
- "Tools for mapping high-throughput sequencing data". Bioinformatics. 28 (24): 3169–77. December 2012. doi:10.1093/bioinformatics/bts605. PMID 23060614. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "TopHat: discovering splice junctions with RNA-Seq". Bioinformatics. 25 (9): 1105–11. May 2009. doi:10.1093/bioinformatics/btp120. PMID 19289445. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation". Nature Biotechnology. 28 (5): 511–5. May 2010. doi:10.1038/nbt.1621. PMID 20436464. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Assembly algorithms for next-generation sequencing data". Genomics. 95 (6): 315–27. June 2010. doi:10.1016/j.ygeno.2010.03.001. PMID 20211242. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Assessing De Novo transcriptome assembly metrics for consistency and utility". BMC Genomics. 14: 465. July 2013. doi:10.1186/1471-2164-14-465. PMID 23837739. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "TransRate: reference-free quality assessment of de novo transcriptome assemblies". Genome Research. 26 (8): 1134–44. August 2016. doi:10.1101/gr.196469.115. PMID 27252236. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Evaluation of de novo transcriptome assemblies from RNA-Seq data". Genome Biology. 15 (12): 553. December 2014. doi:10.1186/s13059-014-0553-5. PMID 25608678. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Velvet: algorithms for de novo short read assembly using de Bruijn graphs". Genome Research. 18 (5): 821–9. May 2008. doi:10.1101/gr.074492.107. PMID 18349386. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Oases: robust de novo RNA-seq assembly across the dynamic range of expression levels". Bioinformatics. 28 (8): 1086–92. April 2012. doi:10.1093/bioinformatics/bts094. PMID 22368243. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "De novo assembly and analysis of RNA-seq data". Nature Methods. 7 (11): 909–12. November 2010. doi:10.1038/nmeth.1517. PMID 20935650. الوسيط
|CitationClass=تم تجاهله (مساعدة)صيانة CS1: عرض-المؤلفون (link) - "Full-length transcriptome assembly from RNA-Seq data without a reference genome". Nature Biotechnology. 29 (7): 644–52. May 2011. doi:10.1038/nbt.1883. PMID 21572440. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Using the miraEST assembler for reliable and automated mRNA transcript assembly and SNP detection in sequenced ESTs". Genome Research. 14 (6): 1147–59. June 2004. doi:10.1101/gr.1917404. PMID 15140833. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Genome sequencing in microfabricated high-density picolitre reactors". Nature. 437 (7057): 376–80. September 2005. Bibcode:2005Natur.437..376M. doi:10.1038/nature03959. PMID 16056220. الوسيط
|CitationClass=تم تجاهله (مساعدة)صيانة CS1: عرض-المؤلفون (link) - "Comparing de novo assemblers for 454 transcriptome data". BMC Genomics. 11: 571. October 2010. doi:10.1186/1471-2164-11-571. PMID 20950480. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing". Journal of Computational Biology. 19 (5): 455–77. May 2012. doi:10.1089/cmb.2012.0021. PMID 22506599. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome". BMC Bioinformatics. 12: 323. August 2011. doi:10.1186/1471-2105-12-323. PMID 21816040. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Kovaka, Sam; Zimin, Aleksey V.; Pertea, Geo M.; Razaghi, Roham; Salzberg, Steven L.; Pertea, Mihaela (2019-07-08). "Transcriptome assembly from long-read RNA-seq alignments with StringTie2". bioRxiv: 694554. doi:10.1101/694554. مؤرشف من الأصل في 29 نوفمبر 2020. اطلع عليه بتاريخ 27 أغسطس 2019. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Visualization of omics data for systems biology". Nature Methods (باللغة الإنجليزية). 7 (3 Suppl): S56–68. March 2010. doi:10.1038/nmeth.1436. PMID 20195258. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "HTSeq—a Python framework to work with high-throughput sequencing data". Bioinformatics. 31 (2): 166–9. January 2015. doi:10.1093/bioinformatics/btu638. PMID 25260700. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Near-optimal probabilistic RNA-seq quantification". Nature Biotechnology. 34 (5): 525–7. May 2016. doi:10.1038/nbt.3519. PMID 27043002. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "The Sequence Alignment/Map format and SAMtools". Bioinformatics. 25 (16): 2078–9. August 2009. doi:10.1093/bioinformatics/btp352. PMID 19505943. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2". Genome Biology. 15 (12): 550. 2014. doi:10.1186/s13059-014-0550-8. PMID 25516281. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Ballgown bridges the gap between transcriptome assembly and expression analysis". Nature Biotechnology (باللغة الإنجليزية). 33 (3): 243–6. March 2015. doi:10.1038/nbt.3172. PMID 25748911. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Design and validation issues in RNA-seq experiments". Briefings in Bioinformatics. 12 (3): 280–7. May 2011. doi:10.1093/bib/bbr004. PMID 21498551. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "An abundance of ubiquitously expressed genes revealed by tissue transcriptome sequence data". PLOS Computational Biology. 5 (12): e1000598. December 2009. Bibcode:2009PLSCB...5E0598R. doi:10.1371/journal.pcbi.1000598. PMID 20011106. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes". Genome Biology. 3 (7): RESEARCH0034. June 2002. doi:10.1186/gb-2002-3-7-research0034. PMID 12184808. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Nascent RNA sequencing reveals widespread pausing and divergent initiation at human promoters". Science. 322 (5909): 1845–8. December 2008. Bibcode:2008Sci...322.1845C. doi:10.1126/science.1162228. PMID 19056941. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Molecular mechanisms of ethanol-induced pathogenesis revealed by RNA-sequencing". PLOS Pathogens. 6 (4): e1000834. April 2010. doi:10.1371/journal.ppat.1000834. PMID 20368969. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Identification and functional validation of a unique set of drought induced genes preferentially expressed in response to gradual water stress in peanut". Molecular Genetics and Genomics. 281 (6): 591–605. June 2009. doi:10.1007/s00438-009-0432-z. PMID 19224247. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Tavassoly, Iman; Goldfarb, Joseph; Iyengar, Ravi (2018-10-04). "Systems biology primer: the basic methods and approaches". Essays in Biochemistry (باللغة الإنجليزية). 62 (4): 487–500. doi:10.1042/EBC20180003. ISSN 0071-1365. PMID 30287586. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "RNA-Seq and human complex diseases: recent accomplishments and future perspectives". European Journal of Human Genetics. 21 (2): 134–42. February 2013. doi:10.1038/ejhg.2012.129. PMID 22739340. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Role of non-coding sequence variants in cancer". Nature Reviews Genetics. 17 (2): 93–108. February 2016. doi:10.1038/nrg.2015.17. PMID 26781813. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Transposable elements and the epigenetic regulation of the genome". Nature Reviews Genetics. 8 (4): 272–85. April 2007. doi:10.1038/nrg2072. PMID 17363976. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Single-cell technologies to study the immune system". Immunology. 147 (2): 133–40. February 2016. doi:10.1111/imm.12553. PMID 26551575. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Translating RNA sequencing into clinical diagnostics: opportunities and challenges". Nature Reviews Genetics. 17 (5): 257–71. May 2016. doi:10.1038/nrg.2016.10. PMID 26996076. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Discovery of virulence factors of pathogenic bacteria". Current Opinion in Chemical Biology. 12 (1): 93–101. February 2008. doi:10.1016/j.cbpa.2008.01.023. PMID 18284925. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Prediction of antibiotic resistance by gene expression profiles". Nature Communications. 5: 5792. December 2014. Bibcode:2014NatCo...5.5792S. doi:10.1038/ncomms6792. PMID 25517437. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Dual RNA-seq of pathogen and host" (PDF). Nature Reviews. Microbiology. 10 (9): 618–30. September 2012. doi:10.1038/nrmicro2852. PMID 22890146. مؤرشف من الأصل (PDF) في 29 نوفمبر 2020. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "A review on computational systems biology of pathogen-host interactions". Frontiers in Microbiology. 6: 235. 2015. doi:10.3389/fmicb.2015.00235. PMID 25914674. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Transcriptome analyses reveal genotype- and developmental stage-specific molecular responses to drought and salinity stresses in chickpea". Scientific Reports. 6: 19228. January 2016. Bibcode:2016NatSR...619228G. doi:10.1038/srep19228. PMID 26759178. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Candida albicans biofilms: a developmental state associated with specific and stable gene expression patterns". Eukaryotic Cell. 3 (2): 536–45. April 2004. doi:10.1128/EC.3.2.536-545.2004. PMID 15075282. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "The origin of malignant malaria". Proceedings of the National Academy of Sciences of the United States of America. 106 (35): 14902–7. September 2009. Bibcode:2009PNAS..10614902R. doi:10.1073/pnas.0907740106. PMID 19666593. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Drug resistance. Population transcriptomics of human malaria parasites reveals the mechanism of artemisinin resistance". Science. 347 (6220): 431–5. January 2015. Bibcode:2015Sci...347..431M. doi:10.1126/science.1260403. PMID 25502316. الوسيط
|CitationClass=تم تجاهله (مساعدة)صيانة CS1: عرض-المؤلفون (link) - "Molecular mechanisms of metal hyperaccumulation in plants". The New Phytologist. 181 (4): 759–76. March 2009. doi:10.1111/j.1469-8137.2008.02748.x. PMID 19192189. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "RNA-Seq improves annotation of protein-coding genes in the cucumber genome". BMC Genomics. 12: 540. November 2011. doi:10.1186/1471-2164-12-540. PMID 22047402. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "A transcriptome resource for the koala (Phascolarctos cinereus): insights into koala retrovirus transcription and sequence diversity". BMC Genomics. 15: 786. September 2014. doi:10.1186/1471-2164-15-786. PMID 25214207. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "A SNP resource for Douglas-fir: de novo transcriptome assembly and SNP detection and validation". BMC Genomics. 14: 137. February 2013. doi:10.1186/1471-2164-14-137. PMID 23445355. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "De novo transcriptome assembly for the lobster Homarus americanus and characterization of differential gene expression across nervous system tissues". BMC Genomics. 17: 63. January 2016. doi:10.1186/s12864-016-2373-3. PMID 26772543. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "A transcriptome based aging clock near the theoretical limit of accuracy". bioRxiv. 2020. doi:10.1101/2020.05.29.123430. مؤرشف من الأصل في 29 نوفمبر 2020. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Predicting age from the transcriptome of human dermal fibroblasts". Genome Biology. 19 (1): 221. 2018. doi:10.1186/s13059-018-1599-6. PMID 30567591. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Ribosomal RNA and translation". Annual Review of Biochemistry. 60: 191–227. 1991. doi:10.1146/annurev.bi.60.070191.001203. PMID 1883196. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Functional requirement of noncoding Y RNAs for human chromosomal DNA replication". Molecular and Cellular Biology. 26 (18): 6993–7004. September 2006. doi:10.1128/MCB.01060-06. PMID 16943439. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "The snoRNA HBII-52 regulates alternative splicing of the serotonin receptor 2C". Science. 311 (5758): 230–2. January 2006. Bibcode:2006Sci...311..230K. doi:10.1126/science.1118265. PMID 16357227. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Non-coding RNAs: hope or hype?". Trends in Genetics. 21 (5): 289–97. May 2005. doi:10.1016/j.tig.2005.03.007. PMID 15851066. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Non-coding RNAs in human disease". Nature Reviews Genetics. 12 (12): 861–74. November 2011. doi:10.1038/nrg3074. PMID 22094949. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Gene Expression Omnibus". www.ncbi.nlm.nih.gov. مؤرشف من الأصل في 29 نوفمبر 2020. اطلع عليه بتاريخ 26 مارس 2018. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Minimum information about a microarray experiment (MIAME)-toward standards for microarray data". Nature Genetics. 29 (4): 365–71. December 2001. doi:10.1038/ng1201-365. PMID 11726920. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "ArrayExpress update—simplifying data submissions". Nucleic Acids Research. 43 (Database issue): D1113–6. January 2015. doi:10.1093/nar/gku1057. PMID 25361974. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Minimum Information About a Microarray Experiment (MIAME)--successes, failures, challenges". TheScientificWorldJournal. 9: 420–3. May 2009. doi:10.1100/tsw.2009.57. PMID 19484163. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Expression Atlas update—an integrated database of gene and protein expression in humans, animals and plants". Nucleic Acids Research. 44 (D1): D746–52. January 2016. doi:10.1093/nar/gkv1045. PMID 26481351. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Genevestigator v3: a reference expression database for the meta-analysis of transcriptomes". Advances in Bioinformatics. 2008: 420747. 2008. doi:10.1155/2008/420747. PMID 19956698. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "BodyParts3D: 3D structure database for anatomical concepts". Nucleic Acids Research. 37 (Database issue): D782–5. January 2009. doi:10.1093/nar/gkn613. PMID 18835852. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "NONCODE 2016: an informative and valuable data source of long non-coding RNAs". Nucleic Acids Research. 44 (D1): D203–8. January 2016. doi:10.1093/nar/gkv1252. PMID 26586799. الوسيط
|CitationClass=تم تجاهله (مساعدة)
ملاحظات
- In molecular biology, hybridisation is a phenomenon in which single-stranded deoxyribonucleic acid (حمض نووي ريبوزي منقوص الأكسجين) or ribonucleic acid (حمض نووي ريبوزي) molecules الديناميكا الحرارية للحمض النووي to نيوكليوتيد مكمل.
- One picolitre is about 30 million times smaller than a drop of water.
قراءة متعمقة
- "Transcriptomics technologies". PLOS Computational Biology. 13 (5): e1005457. May 2017. Bibcode:2017PLSCB..13E5457L. doi:10.1371/journal.pcbi.1005457. PMID 28545146. الوسيط
|CitationClass=تم تجاهله (مساعدة) - تحليل النسخ المقارن في الوحدة المرجعية في علوم الحياة
- البرمجيات المستخدمة في النسخ:
 بوابة علم الأحياء
بوابة علم الأحياء