متلازمة خلل التنسج النخاعي
متلازمة خلل التنسج النقوي أو متلازمة خَلَلُ التَّنَسُّجِ النُّخاعِيّ[1] أو متلازمة الثدن النقوي (بالإنكليزية Myelodysplastic Syndrome) عبارة عن نمو غير طبيعي لخلايا الدم في مرحلة التكون النخعي.
كانت تعرف في الماضي باسم -مُقَدِّماتُ الابْيِضاض- وهي حالة طبية تنطوي على إنتاج خلايا دم نقويه غير فعالة. يتصف المرض بأنه مجموعة من الأعراض التي تؤدي إلى هبوط كل من كريات الدم الحمراء والبيضاء والصفائح الدموية. تحدث الإصابة غالبا عند البالغين ونادرا حدوثها عند الأطفال أو تحت سن الخمسين، ومعدل عمر المريض عند الإصابة ما بين 65-75 عاما.
غالباً ما يصبح لدى المرضى المصابين بهذه المتلازمه فقر دم حاد، ويحتاجون لنقل دم متكرر. وفي معظم الحالات يتفاقم المرض، ويصاب المريض بـ قلة الكريات وذلك بسبب الفشل المُطرِد لنخاع العظم. وتتحول المتلازمة لدى ثلث المرضى إلى مرض سرطاني خبيث يسمى ابيضاض الدم النقوي الحاد عادة خلال أشهر إلى بضع سنوات.
متلازمة خلل التنسج النقوي هي جميع اضطرابات الخلايا الجذعية المكونة للدم داخل نخاع العظم، حيث تتكون خلايا الدم بشكل مضطرب وغير فعال، وبذلك تنخفض كمية ونوعية الخلايا المكونة للدم بشكل غير انعكاسي-لارجعة فيه-.
| متلازمة خلل التنسج النقوي | |
|---|---|
| معلومات عامة | |
| الاختصاص | علم الدم، علم الأورام |
| من أنواع | مرض تكاثر نقوي |
| الإدارة | |
| أدوية | فيرونوستات ، وديفيروكسامين ، وإيبويتين ألفا ، وديفيريبرون ، وفيلغراستيم ، ونيفولوماب ، وليناليدوميد ، وسيكلوسبورين ، وديسيتابين ، وسيتارابين ، وروميبلوستيم ، وأزاسيتيدين ، وإلترومبوباغ ، وروكسوليتينيب ، وإيبيليموماب ، وديفيراسيروكس ، وفينيتوسلاكس ، وثاليدوميد
|
العلامات والأعراض
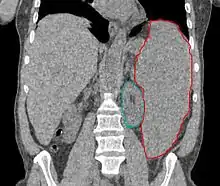
يكون متوسط عمر المريض عند تشخيص المتلازمة بين 60 و 75 سنة، قليل من المرضى يتم تشخيص المتلازمة لديهم تحت سن 50 سنة، اما الأطفال فمن النادر اصابتهم بالمتلازمة، كما ان أصابة الذكور بالمرض أكثر شيوعاً من الإناث، أعراض وعلامات المرض غير محددة، ومتعلقة بشكل عام بقلة كريات الدم:
- فقر دم-إعياء مزمن، ضيق التنفس، الإحساس بالبرودة، وفي بعض الأحيان ألم الصدر.
- قلة العدلات-زيادة احتمالية العدوى
- قلة الصفيحات-زيادة القابلية للنزف وللكدمات(الرضوض) والنزف تحت الجلد مما يؤدي إلى حصول الفرفرية-طفح جلدي نزفي- أو الحبر
العديد من الأفراد لايوجد لديهم أعراض للمرض، ويتم تحديد حالة قلة الكريات والمشاكل الآخرى عن طريق فحص الدم الروتيني:
- قلة العدلات، فقر الدم وقلة الصفيحات، على التوالي!
- تضخم الطحال أو تضخم الكبد في النادر
- حبيبات غير طبيعية داخل الخلايا، شكل وحجم النواة غير طبيعي
- وتشوهات صبغية، بما في ذلك إزفاء الكروموسومات وأعداد غير طبيعية للكروموسومات
برغم وجود نسبة خطورة للإصابة بابيضاض الدم النقوي الحاد إلا أن 50% من الوفيات تحدث نتيجة النزف أو العدوى.
أبيضاض الدم الذي يحدث نتيجة خلل التنسج، لوحظ مقاومته للعلاج.
فيزيولوجيا المرض
متلازمة خلل التنسج النقوي تنجم من التعرض لعوامل بيئية مثل الإشعاعات ومادة البنزين، بالإضافة لبعض العوامل الأخرى التي يتم الإبلاغ عنها بشكل متقطع.
حالات خلل التنسج النقوي الثانوي، تحدث لدى مرضى السرطان كنتيجة للمرحلة المتأخرة من التسمم العلاجي لمرض السرطان، والتركيبة العلاجية عادةً ماتكون مؤلفة من الإشعاعات والعامل المؤلكل محاكي الإشعاع، مثل: بوسلفان، نيتروسويريا، أو بروكاربازين خلال مدة 5 - 7 سنوات، أو مثبطات التوبوأيزومراز في الحمض النووي لمدة سنتين.
حالات فقر الدم اللاتنسجي المكتسب (نتيجةً للعلاجات المثبطة للمناعة) وفقر دم فانكوني، كلا الحالتين من الممكن أن تتطور إلى متلازمة خلل التنسج النقوي.
تنشأ متلازمة خلل التنسج النقوي من طفرة حصلت في الخلايا الجذعية المكونة للدم أدت إلى ظهور المرض، لكن تظل نوعية العيوب المسئولة عن هذه الأمراض غير مفهومة بالشكل الكافي.
التمايز الخلوي في الخلايا السلائف ضعيف، وهناك ازدياد واضح في عدد الخلايا الميتة بالموت المبرمج بين خلايا النخاع العظمي، كما أن التوسع النسيلي للخلايا الغير طبيعية يؤدي إلى إنتاج خلايا ليس لديها قدرة على التمايز، واذا زادت النسبة الإجمالية لأرومات النخاع العظمي (فوق 20 - 30%) عندها يكون التحول لابيضاض الدم النقوي الحاد ميالاً للحدوث. وهذا خير مثال لنظرية الخطوات المتعددة للتسرطن -فرضية ندسون- حيث يؤدي حدوث عدة طفرات في الخلايا الطبيعية إلى تحولها لسرطان. يجدر القول أن نسبة الإعاقة والوفيات المنسوبة للمتلازمة، تنجم عن الضرر الذي يلحق المريض نتيجة قلة الكريات -المشاهد في جميع المرضى- وليس بالضرورة نتاج التحول الخبيث إلى أبيضاض الدم النقوي الحاد، لذلك يحتاج المريض لنقل دم باستمرار لمعالجة فقر الدم الناجم عن قلة الكريات، وهو الأمر الذي يسبب اكتناز الحديد للمريض، وذلك إذا استمرت عملية نقل الدم للمريض على المدى الطويل، بالإضافة إلى حدوث اثنين من المضاعفات الخطيرة التي تحدث للمرضى نتيجة قلة الكريات:
- النزف -نتيجة قلة الصفيحات-
- أو العدوى -نتيجة قلة الكريات البيض
الوراثيات
التعرف على التغيرات الجينية في الحمض النووي -تغيرات التخلق المتوالي- في متلازمة خلل التنسج النقوي يفسر نجاح اثنين من أصل ثلاثة أدويه متوفرة تجارياً، وأقرتها إدارة الغذاء والدوء لعلاج المتلازمة.
المَثْيَلَة -إضافة مجموعة ميثيل- المناسبة للحمض النووي تعتبر عملية حرجة وحساسة في تنظيم الجينات خلال التكاثر، وقد يؤدي فقد السيطرة على عملية المَثْيَلَة إلى نمو الخلايا بدون تحكم، وقلة الكريات، إلا أن أحد الأدوية المُقَرة حالياً من إدراة الغذاء والدواء، تعطي نتائج ايجابية أكثر في تنظيم عملية المَثْيَلة داخل نواة الخلايا الجذعية المكونة للدم، وتأثيرها العلاجي يؤدي إلى استرجاع التعداد الطبيعي للدم، كما يؤدي لتقليل فرص التحول لـ ابيضاض الدم الحاد، عن طريق تأخير التفاقم المرضي لمتلازمة خلل التنسج النقوي.
بعض الباحثين اقترح فكرة ربما تفسر كيفية حدوث المتلازمة، مفادها: تراكم الطفرات الجينية في الخلايا الجذعية المكونة للدم، الناتج -بمرور الوقت- عن القصور الوظيفي للميتوكوندريا، وهذا يمثل زيادة في نسبة حدوث المرض بين المسنين، وقد عزى الباحث هذا الاقتراح إلى أن تراكم حديد الميتوكوندريا المترسب داخل أرومات الحديد الحلقية، يعطي دليلاً على القصور الوظيفي للميتوكوندريا في متلازمة خلل التنسج النقوي..
متلازمة 5q-
منذ مالا يقل عن سنة 1974، ارتبط حذف الذراع الطويل من الكروموسوم 5 بتشوهات الخلايا الجذعية المكونة للدم، بحلول عام 2005 تم تسجيل فاعلية دواء ليناليدوميد مع مرضى متلازمة خلل التنسج النقوي المتعلقة بمتلازمة شطب كروموسوم 5 وفي شهر ديسمبر من نفس العام أقرته إدارة الغذاء والدواء لهذه الحالة.
التشخيص
يجب التفريق بين فقر الدم وقلة الصفيحات و/أو نقص الكريات البيضاء، عند تشخيص المتلازمة.
الفحوصات الاعتيادية تشمل:
- عد دموي شامل واختبار فلم الدم، اختبار فلم الدم قد يعطي قرائن تثبت وجود تكسر في خلايا الدم، أو تلازن في صفائح الدم، تؤدي إلى قلة الصفيحات الكاذبة أو ابيضاض الدم
- اختبارات الدم، لاستبعاد اي أسباب أخرى ممكن أن تؤدي لقلة الكريات مثل: الذئبة الحمامية، الالتهاب الكبدي، نقص فيتامين بي 12 أو فيتامين بي 9 وغيره من الفيتامينات، قصور كلوي، قصور القلب، فيروس الايدز، فقر الدم الانحلالي، اعتلال غامائي أحادي النسيلة.
- فحص نخاع العظم بواسطة علماء أمراض الدم.
- الوراثِيَات الخلوية -دراسة الصبغ الوراثية-
- قياس التدفق الخلوي، تساعد في مراقبة اي خلل في تكاثر الخلايا اللمفاوية في النخاع.
في البداية تظهر أعراض فقر الدم، معظم المرضى يَشْكُون بدايات تدريجية من الشعور بالإعياء والضعف، ضيق النفس والشحوب، إلا أن النصف من المرضى على الاقل لايَشْكُون من أعراض، ويكون أكتشاف المتلازمة لديهم مصادفة، وبشكل عرضي أثناء إجراء اختبارت الدم الروتينية، أيضا من الضروري معرفة جانب من تاريخ المريض، إذا ما كان قد تعرض لإشعاعات في حياته أو كان تحت علاج كيميائي مسبق.
وجود الحرارة وفقدان للوزن يشير إلى احتمالية وجود مرض التكاثر النقوي أكثر من احتمالية الإصابة بالمتلازمة.
- كما أن الأطفال الذين لديهم منغوليا معرضين للإصابة بالمتلازمة، ربما يكون هناك فقر الدم الحديدي الأرومات أو فقر دم فانكوني.
متوسط عمر الفرد عند تشخيص متلازمة خلل التنسج النقوي يكون قرابة 65 سنة، لكن هناك حالات خلل تنسج مسجلة لدى الأطفال، بعض المرضى لديهم تاريخ علاجي بالكيماويات، خصوصاً -العوامل المؤلكلة- مثل: سايكلوفوسفاميد، ميلفالان، بوسلفان وكلورامبيوسيل، والبعض الآخر يكون قد تعرض للإشعاعات (إما لغرض علاجي أو عن طريق الخطأ) أو الاثنين معاً، على سبيل المثال: أثناء نقل خلايا جذعية لعلاج مرض اخر، وزيادة تعرض عمال المصانع للهيدروكربونات -المحروقات- مثل مصانع النفط، لديهم نسبة قليلة من الخطورة أكثر من عامة السكان. كذلك الذكور أكثر قليلاً تأثرا بالمرض من الإناث.التعرض للبنزين والزايلين له علاقة بالتلازمة، المحاربين الفتناميين القدامى الذين سبق لهم التعرض للعامل البرتقالي لديهم نسبة خطورة للإصابة بالمتلازمة..
العلاج
الغرض من علاج حالات خلل التنسج، هو تخفيف أعراض المرض، استرجاع الحيوية، واطالة مدة البقاء، والتقليل من احتمالية الإصابة بأبيضاض الدم النقوي الحاد.
- الأدوية:أقرت إدارة الأغذية والأدوية ثلاثة أدوية لعلاج متلازمة خلل التنسج النقوي:
- ازاسيتيدين (بالإنجليزية: Azacitidine): متوسط البقاء 21 شهر.
- ديسيتابين: (بالإنجليزية: Decitabine) 43% هي أعلى استجابة كاملة مسجلة.
- ليناليدوميد: يوصف لعلاج متلازمة خلل التنسج، المتعلقة بمتلازمة حذف الكروموسوم5 -مجموعة فرعية من متلازمة خلل التنسج النقوي- له تأثير فعال في الحد من احتياج المريض لخلايا الدم الحمراء المنقولة اليه.
- العلاج الكيميائي مع تقليل عوامل المثيلة.
- زراعة نخاع العظم.
- علاج اكتناز الحديد المتسبب من تكرار نقل الدم للمريض.
- التجربة سريرية
توقعات سير المرض
مؤشرات المسار الجيد للمرض: صغر السن: نقص طبيعي أو متوسط في العدلات أو الصفائح وتعداد منخفض للأرومات داخل نخاع العظم (أقل من 20%)، لاوجود للأرومات في تيار الدم، لاوجود لأجسام آور، أرومة الحديد الحلقية، نمط نووي طبيعي لمختلف الأنماط النووية وبدون شذوذ في المركبات الصبغية، وعند زرع النخاع في المختبر، لاوجود لنمط نمو ابيضاضي.
مؤشرات المسار الضعيف: جميع المؤشرات المقابلة للمسار الجيد: التقدم في السن: قلة العدلات أو الصفيحات الشديد، كثرة الأرومات داخل نخاع العظم (20 - 29%) أو في تيار الدم، وجود اجسام آور، غياب أرومة الحديد الحلقية، موقع غير طبيعي أو سلائف محببات غير ناضجة في جزء من النخاع العظمي، أنماط نووية غير طبيعية، وتشوهات في مركبات الصبغة الوراثية للنخاع، وجود نمط نمو لإبيضاض الدم في مزرعة النخاع.
الوبائيات
لايعرف على وجه التحديد عدد الأفراد الذين لديهم متلازمة خلل التنسج، لوجود بعض الحالات الغير مُشَخصه، بعض التقديرات تتحدث عن قرابة 10,000الى20,000 الف حالة جديدة سنوياً في الولايات المتحدة فقط، وربما تتزايد نسبة حدوث المرض بازدياد أعمار السكان، ويقترح بعض الباحثين ارتفاع نسبة حدوث المرض بين المرضى فوق السبعين سنة بمعدل 15 حالة لكل 100,000 شخص، سنوياً.
متلازمة خلل التنسج النقوي (خَلَلُ التَّنَسُّجِ النُّخاعِيّ) أو متلازمة الثدن النقوي (بالإنكليزية Myelodysplastic Syndrome) حالة مرضيّة مرتبطة بالإنتاج غير الفعّال لخلايا الدم أو بخلل في تنسُّج (تكوّن أنسجة) هذه الخلايا.[2]
قد تؤدي متلازمة خلل التنسج النقوي إلى حاجة المريض لنقل وحدات الدم له أو إصابته بفقر الدم (الأنيميا). في بعض الحالات قد تصل الأمور إلى ما هو أسوأ من ذلك عندما ينخفض تعداد كريات الدم نتيجة اعتلال نخاع العظم. تعتمد الأعراض في متلازمة خلل التنسّج على نوع وشدّة المرض؛ يعيش الكثير من المرضى حياةً طبيعية، غالبًا لا يعلم المريض بإصابته ولا تظهر عليه أي أعراض حتى يكتشف إصابته من خلال فحص دم روتينيّ.
تعرف متلازمة خلل التنسّج بأنها أي خلل مرتبط في الخلايا الجذعية للدم في نخاع العظم. حيث يكون إنتاج خلايا الدم غير فعّال. بمرور الوقت تنخفض نوعية وعدد خلايا الدم إلى غير رجعة، لتصل فيما بعد إلى توقف إنتاج خلايا الدم.[3] متوسط عمر الإصابة بالمرض هو 68 عامًا.
التصنيف
التصنيف الفرنسي- الأمريكي- البريطاني FAB
بين عامي 1974 و 1975 قام مجموعة من علماء الأمراض من فرنسا، الولايات المتحدة وبريطانيا بوضع أول تصنيف لهذه الأمراض. نُشر هذا التصنيف عام [4] 1976 ونُقّح عام 1982. استخدم من قبل علماء الأمراض والأطباء على مدى 20 عامًا. صنّفت الأمراض وفقًا لهذا النظام إلى خمس فئات:
| التصنيف العالمي للأمراض المرتبطة بعلم الأورام ICD-O | الاسم | التوصيف |
|---|---|---|
| M9980/3 | فقر الدم الحرون RA | تتصف هذه الحالة بوجود خلايا الدم البدائية (الأرومة النقوية) بنسبة أقل من 5% في نخاع العظم، بالإضافة إلى خلل مرضي في خلايا الدم الحمراء الأم. |
| M9982/3 | فقر الدم الحرون مع الأرومات الحديدية الحلقية RASA | تتصف هذه الحالة أيضًا بوجود خلايا الدم البدائية (الأرومة النقوية) بنسبة أقل من 5% في نخاع العظم، كما تظهر فيها بنسبة 15% أو أكثر خلايا الدم الحمراء الأم في نخاع العظم على شكل أرومات حديدية حلقية(خلايا ممتلئة بعنصر الحديد على شكل حلقات بدلا من شكلها الطبيعي القرصي). |
| M9983/3 | فقر الدم الحرون مع فيض الأرومات RAEB | تتصف بوجود أرومات نقوية في نخاع العظم بنسبة 5-20%. |
| M9984/3 | فقر الدم الحرون مع فيض الأرومات المتحولة RAEB-T | تتصف بوجود أرومات نقوية في نخاع العظم بنسبة 21-30% (إذا وصلت نسبة الأرومات أكثر من 30% تعرف الحالة بأنها ابيضاض الدم النقوي الحاد). |
| M9945/3 | الابيضاض الوحيدي النقوي المزمن CMML (يجب تمييزه عن ابيضاض الدم النقوي المزمن)* | يتصف بوجود أرومات نقوية في نخاع العظم بنسبة أقل من 20%، كما أن نوع من خلايا الدم البيضاء تعرف بـ (الوحيدة monocyte) تتواجد بتركيز أعلى من 1*109 لكل لتر في الدم الطرفي. |
(بإمكانكم الرجوع لجدول يقارن هذين المرضين على Cleveland Clinic.[5])
يعتبر فقر الدم الحرون RA وفقر الدم الحرون مع الأرومات الحديدية الحلقية RARS أفضل حالتين من حيث شدة المرض؛ حيث أن متوسط ما يعيشه الشخص مع المرض من 3 إلى 5 سنوات، وقد يعيش أكثر من 10 سنوات مع المرض حتى بدون أن تجرى له عملية زراعة نخاع العظم، في حال أجريت له العملية بنجاح يمكن للإنسان أن يصمد أكثر من ذلك. أسوأ حالة على الإطلاق نجدها في فقر الدم الحرون مع فيض الأرومات المتحولة RAEB-T حيث يبلغ متوسط عمر المريض أقل من عام واحد. في ربع الحالات ينتهي المرضى بابيضاض دم واضح، يموت الآخرون نتيجة مضاعفات انخفاض تعداد كريات الدم أو أمراض أخرى. النظام العالمي الإنذاري أو النظام العالمي لرصد المآل The International Prognostic Scoring System هو نظام آخر لتشخيص متلازمة خلل التنسج النخاعي نشر في الدم عام 1997،[6] يأخذ هذا النظام في عين الاعتبار نسبة الأرومات في نخاع العظم، الدراسات الجينية لخلايا الدم وتعداد كريات الدم.
منظمة الصحة العالمية
في أواخر التسعينات قام مجموعة من علماء الأمراض والأطباء الذين يعملون تحت راية منظمة الصحة العالمية بتطوير نظام التصنيف هذا؛ بإضافة مجموعة من الأمراض واستثناء أمراض أخرى، قامت منظمة الصحة العالمية في 2008 بتطوير خطة جديدة تستند إلى البيانات الجينية. إلا أن دراسة شكل خلايا الدم (morphology) في الدم الطرفي ودراسة خزعات نخاع العظم مازالت هي المعيار المعتمد في تحديد أفضل تصنيف للخلل في تعداد خلايا الدم.
| النظام القديم | النظام الحديث |
|---|---|
| فقر الدم الحرون RA | قلّة الكريات الحرون مع الثدن أحادي السلالة (فقر الدم الحرون، قلة العدلات الحرون وقلّة كريات الدم الحمراء الحرون). |
| فقر الدم الحرون مع الأرومات الحديدية الحلقية RASA | فقر الدم الحرون مع الأرومات الحديدية الحلقية- كثرة الصفائح (RARS-t) (كيان مؤقت ) والذي هو بالضرورة اعتلال خلل التنسج النقوي/ تكاثري نقوي وله عادةً طفرة إنزيم ثنائي الوجه JAK2- بحسب التصنيف الجديد لمنظمة الصحة العالمية. |
| قلة الكريات الحرون مع الثدن متعدد السلالات (RCMD) يتضمن قلة الكريات الحرون مع الثدن متعدد السلالات والأرومات الحديدية الحلقية (RCMD-RS). يقع تحت فئة RCMD المرضى الذين عندهم تغيرات مرضية غير مقتصرة على خلايا الدم الحمراء؛ بمعنى آخر ثدن خلايا الدم البيضاء الأم البارزة وثدن الصفائح الأم. | |
| فقر الدم الحرون مع فيض الأرومات RAEB | فقر الدم الحرون مع فيض الأرومات تم تقسيمه إلى نوعين: RAEB-I (نسبة الأرومات 5-9%) و RAEB-II (نسبة الأرومات 10-19%). توقعات سير النوع الثاني من المرض للتحسن أو الشفاء هي أقل منها للأول. كما أن أجسام آور يمكن رؤيتها في النوع الثاني وهو ما يجعل تمييزه عن ابيضاض الدم النقوي الحاد أمرًا صعبًا. |
| فقر الدم الحرون مع فيض الأرومات المتحولة RAEB-T | لقد تم استثناء هذه الفئة، المرضى الذين يتبعون لها يعتبرون أنهم مصابون بابيضاض الدم النقوي الحاد.
تمت إضافة متلازمة 5q؛ تُشاهد عادةً عند النساء الأكبر سنًا اللواتي يمتلكن تعداد صفائح عالٍ أو عاديّ بالإضافة إلى الحذوفات المعزولة للذراع الطويل لكروموسوم 5 في خلايا نخاع العظم لهذا التصنيف. |
| الابيضاض الوحيدي النقوي المزمن CMML | تمّ حذفه من هذا تصنيف متلازمة خلل التنسج النقوي وإضافته إلى تصنيف المتلازمات الناتجة عن تقاطع خلل التنسج النقوي- الخلل التكاثري النقوي. |
| خلل التنسج النقوي غير الخاضع للتصنيف (يمكن رؤيته في حالات ثدن النّوّاء مع التليّف وغيره) | |
| نقص الكريات الحرون عند الأطفال (الثدن في مرحلة الطفولة) دخل حديثًا (عام 2008) إلى تصنيف منظمة الصحة العالمية. |
لم يُجمع الأطباء على هذا التصنيف الحديث وذلك بسبب عدم الحصول على صورة كافية ومفهومة عن فيزيولوجيا هذه الأمراض وفهمها بشكل كامل.
متلازمة خلل التنسج النخاعي غير الخاضعة للتصنيف
بالرغم من أن منظمة الصحة العالمية قامت بوضع تصنيف يغطّي أنواع متلازمة خلل التنسّج النخاعي إلّا أن هناك حالات بقيت عصيّة على التصنيف بسبب خصائص غير اعتيادية:
- في حالات نادرة تكون نسبة الأرومات أقل من 5% مع قضبان آور. هذه الحالات تماثل في خصائصها فقر الدم الحرون مع خلل التنسج متعدد الأرومات RAMD.
- حالات خلل التنسّج النقوي الموضعية تظهر مع قلة العدلات أو قلة الصفيحات بدون فقر الدم ويكون خلل التنسج مقتصراً على أرومة-سلالة- واحدة. يستخدم أحيانًا مصطلح قلة العدلات الحرون- و-قلة الصفيحات الحرون لوصف هذه الحالات التي يجب أن تشخّص بحذر.
- مرضى فقر الدم الحرون RA وفقر الدم الحرون مع فيض الأرومات RAEB قد تصاحبهم كثرة الصفيحات أو كثرة كريات الدم البيضاء بدلًا من انخفاض تعداد خلايا الدم المعتاد.
العلامات والأعراض
إن متوسط عمر تشخيص الإصابة بمتلازمة خلل التنسج النخاعي يتراوح بين 60 و 75 عامًا؛ حالات قليلة شُخّصت تحت ال 50 ونادرًا ما يشخص المرض عند الأطفال. تزيد قليلًا احتمالية الإصابة عند الرجال عنها عند النساء. العلامات والأعراض غير محددة ومرتبطة إجمالُا بانخفاض تعداد كريات الدم:
- فقر الدم الأنيميا؛ (انخفاض تعداد كريات الدم الحمراء أو انخفاض الهيموجلوبين)، الإعياء المزمن، قصور في التنفس، قشعريرة وأحيانًا ألم في الصدر.
- قلة العدلات (انخفاض عدد العدلات) يزيد قابلية الإصابة بالالتهابات.
- قلة الصفائح (انخفاض تعداد الصفائح الدموية)؛ مما يؤدي لزيادة قابلية النزيف وظهور آثارالكدمات ونزيف تحت الجلد مما ينتج عنه الحبرة والفرفرية.[7]
قد يظهر الكثير من مرضى متلازمة خلل التنسّج النخاعي بلا أعراض المرض أو نقص تعداد كريات الدم وما يتبعه من مشاكل:
- قلة العدلات، فقر الدم الأنيمي وقلة الصفيحات (نقص تعداد كريات الدم البيضاء، الحمراء والصفائح الدموية بالترتيب).
- تضخّم الطحال أو في حالات نادرة تضخّم الكبد.
- حبيبات غير طبيعية في الخلايا، شكل وحجم غير طبيعي للأنوية و/أو اضطرابات كروموسومية مثل الإزفاء(تغير المواقع الجينية على الكروموسومات ) (chromosomal translocations) و عدد الكروموسومات غير الطبيعي.
مع أن هناك خطورة لحدوث ابيضاض حاد نقوي المنشأ إلّا أن 50% من الوفيات تحدث نتيجة النزيف أو الالتهاب. كما أن سرطان الدم (اللوكيميا) الناتج عن متلازمة خلل التنسّج النقوي مقاوم للعلاج.
فيزيولوجيا المرض
إن متلازمة خلل التنسج النقوي قد تكون ناتجة عن التعرّض لظروف بيئية مثل الإشعاعات والبنزين، ولكن لم تثبت بشكل قاطع مسببات هذا المرض. متلازمة خلل التنسج النقوي الثانوية تحدث نتيجة العلاج من السرطان باستخدام الأشعة والعوامل المؤلكلة (القاعدية الشبيهة بالمشعة ) مثل بُوسَلْفان، نِتْروزويُورْيا، بروكاربازين (مع فترة تستر تمتد من 5 إلى 7 سنوات) أو نتيجة مضادات انزيم توبيوميريز الحمض النووي (سنتين). كما أن فقر الدم اللاتنسجي المكتسب الناتجة عن العلاجات المثبطة للمناعة وفقر دم فانكوني (Fanconi's anemia) يمكن أن تؤدي إلى متلازمة خلل التنسج النقوي.
متلازمة خلل التنسج النقوي قد تظهر نتيجة التغيرات (الطفرات ) في خلايا نخاع العظم الجذعية متعددة الفاعلية، ولكن السبب المحدد لهذا النوع من الأمراض ما زال غير مفهوم بشكل كافٍ؛ توقف تمايز خلايا الدم الأم، الارتفاع الملحوظ في معدلات موت الخلايا في نخاع العظم. التوَسُّعٌ النَسيلِيّ للخلايا غير الطبيعية ينتج عنه خلايا غير قادرة على الانقسام. إذا كانت نسبة الأَرومَةُ النِّقَوِيَّة في نخاع العظم أعلى من 20% وفقًا لمنظمة الصحة العالمية أو 30% وفقًا للنظام الفرنسي الأمريكي البريطاني يتحول عندها المرض لابيضاض نقوي حاد. هذا التحول يعتبر مثالًا جيدًا على النظرية التي تطرح حدوث سلسلة من التغيرات على خلية طبيعية لتحولها إلى خلية سرطانية وهي نظرية الخطوات المتعددة للتحول للتسرطن.
رغم أنه تاريخيًا كان الاعتراف بالتحول الناتج عن سرطان الدم مهمًا، إلّا أن نسبة مهمة من الحالات المرضية والوفيات المرتبطة بمتلازمة خلل التنسج النقوي ناتجة عن انخفاض تعداد خلايا الدم الملاحظ عند كل المرضى وليس عن تحول المتلازمة إلى ابيضاض نقوي حاد. نادرًا ما يعاني مرضى خلل التنسج النقوي من فقر دم شديد بالرغم من أنه نتيجة حتمية لانخفاض تعداد خلايا الدم عندهم، وذلك يتطلب عملية نقل الدم لهم. إن أخطر المضاعفات المرتبطة بانخفاض تعداد خلايا الدم عند مرضى المتلازمة تتمثل في النزيف (نتيجة انخفاض عدد الصفائح الدموية) والتعرض للالتهابات (نتيجة انخفاض تعداد كريات الدم البيضاء) كما أن نقل الدم لفترات طويلة يؤدي إلى ارتفاع نسبة الحديد في الدم إلى مستويات اعلى من اللازمة.
علم الجينات
أثبتت دراسة التغيرات الجينية في بناء الحمض النووي DNA لمرضى متلازمة خلل التنسج النقوي نجاح دوائين من ثلاثة أدوية معتمدة من قبل مؤسسة الغذاء والدواء الأمريكية FDA لعلاج المرض. إن ميثلة(عملية إضافة مجموعة الميثل العضوية لتركيب الحمض النووي (DNA مهمة جدًا للتحكم في عملية انتشار الجين، وفقدان التحكم في عملية الميثلة يؤدي إلى نمو غير منظم للخلايا وإلى ارتفاع تعداد خلايا الدم.
تظهر أهمية مثبطات إنزيم ناقلة ميثيل الحمض النووي(الانزيم المسؤول عن إضافة مجموعة الميثل إلى DNA) الموافق عليه مؤخرًا في كونها تنتج تحليل أكثر تنظيمًا لمثيلة الحمض النووي DNA في أنوية خلايا الدم الجذعية مما ينتج عنه تعدادٌ طبيعيٌ لخلايا الدم وتأخيرٌ لسرطان الدم الشديد الناتج عن متلازمة خلل التنسج النقوي.
فسر البعض حدوث المتلازمة عند كبار السن بتوقف عمل المايتوكندريا في الخلايا بمرور الوقت مما يؤدي إلى تراكم طفرات الحمض النووي DNA في خلايا الدم الجذعية. كما أشار باحثون إلى تراكم ترسبات الحديد الناتج من المايتوكندريا في الأَرومَةٌ الحَديدِيَّةٌ الحَلَقِيَّة كدليل على توقف عمل المايتوكندريا عند مرضى متلازمة خلل التنسج النقوي.[8]
5q- syndrome
منذ أواخر 1974 ارتبط حذف الذراع الطويل لكروموسوم 5 بخلل التنسّج في خلايا الدم الجذعية.[9][10] بحلول عام 2005 اتضح أن عقار(دواء) ليناليدوميد كان فعالا عند مرضى متلازمة خلل التنسج النقوي المصابون بمتلازمة 5-q .[11] وفي ديسيمبر 2005 وافقت عليه مؤسسة الغذاء والدواء الأمريكية. أفضل استجابة لليناليدوميد كانت عند مرضى متلازمة 5q المعزولة، والمرضى الذين يتعرضون لخطورة منخفضة وفق نتيجة النظام العالمي لقياس أعراض البروستاتا IPSS والمرضى الذين يحتاجون نقل الدم باستمرار. إن تحسن المرض بعد استخدام هذا العلاج عادةً ما يضمن –في المتوسط- 63 شهرًا إضافيًا لحياة المريض. عقار ليناليدوميد له مفعول ثنائي يتمثل في تخفيض عدد السلالات السرطانية عند مرضى متلازمة 5-q وتحفيز تمايز أفضل للخلايا الاولية لكريات الدم الحمر الصحية عند الناس غير المصابين بمتلازمة 5-q .
التغيرات في عوامل الوصل
تظهر التغيرات في عوامل الوصل في 40-80% من حالات متلازمة خلل التنسّج النقوي، تحديدًا الحالات المرتبطة بالأرومات الحلقية.[12]
التشخيص
يجب تمييز متلازمة خلل التنسج النقوي عن فقر الدم، انخفاض الصفائح وقلة كريات الدم البيضاء، استثناء هذه الأمراض التي ينتج عنها انخفاض تعداد خلايا الدم بالإضافة إلى استثناء خلل تنسّج نخاع العظم ضروري لتشخيص الإصابة بمتلازمة خلل التنسج النقوي.
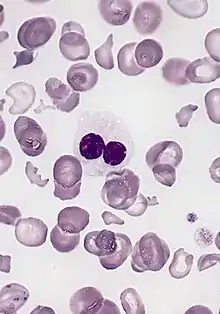
التشخيص التقليدي يتضمن:
- فحص تعداد كريات الدم الكلية وفحص فِلْم الدم. فلم الدم الذي يظهر شكل الخلايا بإمكانه تزويدنا بدلائل فَقْرُ الدَّمِ الانْحِلالِيّ، تجمع الصفائح الدموية يؤدي إلى انخفاض الصفائح الكاذب أو ابيضاض الدم.
- فحوصات دم لاستثناء المسببات الأخرى لانخفاض تعداد خلايا الدم، مثل: الذئبة، التهاب الكبد، نقص B12 أو الفولات (مِلْحُ حَمْضِ الفوليك)، ونقص في فيتامينات أخرى، الفشل الكلوي، فشل عضلة القلب، فَقْرُ الدَّمِ الانْحِلالِيّ، متلازمة نقص المناعة المكتسبة، اعْتِلاَلٌ غامَّائِيّ (خَلَلٌ في تَخَلُّقِ الغلوبولينات المَناعِيَّة) أحادي النسيلة، يجب أيضًا أن يخضع المصابون بفقر الدم لفحوصات السرطان الدورية.
- فحص نخاع العظم. هذا ضروري للتأكد من صحة التشخيص كون نخاع العظم المصاب بخلل التنسّج يعتبر العلامة المميزة لخلل التنسّج النقوي.
- الوِراثِيَّاتُ الخَلَوِيَّة أو الدراسات الكروموسومية. يتم إجراء هذا الفحص في رُشافة نخاع العظم. فحص الوِراثِيَّاتُ الخَلَوِيَّة الاعتيادي يتطلب جمع نماذج حيّة كون الخلايا الحية تدخل طور التقابل في الانقسام الخلوي الخيطي بهدف إظهار الكروموسومات.
- فحص التهجين الموضعي المشع FISH يطلب مرافقًا لفحص الوراثيات الخلوية الاعتيادي، تحدد هذه الفحوصات بسهولة عدد من الاختلالات الكروموسومية المرتبطة بمتلازمة خلل التنسج النقوي وتتضمن del 5q، -7، +8،(تعني حذف قطع جينية من الذراع الطويلة للكروموسوم 5) del 20q(تعني حذف قطع جينية من الذراع الطويلة للكروموسوم رقم 20).
- التَنْميطٌ النَوَوِيّ الافتراضي، يمكن إجراؤه لمرضى متلازمة خلل التنسج النقوي الذين يستخدمون أدوات حسابية لبناء تنميط نووي من الحمض النووي المعطل. هذه الطريقة لا تتطلب زراعة خلايا وتتميز بوضوح أعلى من فحص الوراثيات الخلوية الاعتيادي، ولكن لا يمكنها تحديد الإزفاء(انتقال القطع الجينية ) المتوازن.[13]
- عدّ الكريات المُنساب يساعد في تحديد وجود خلل تكاثُرِيٌّ لِمْفِيّ في نخاع العظم.
- فحص نقص عنصر النحاس يمكن أن يظهر متلازمة خلل التنسج النقوي في فحوصات نخاع العظم.[14]
غالبًا ما يسود فقر الدم (الأنيميا) في المراحل الأولى من المرض. معظم المرضى الذين تظهر عليهم الأعراض يشتكون من احساس يزداد تدريجيا بالإعياء والتعب، ضيق التنفس والشحوب، ولكن نصف المرضى على الأقل لا تظهر عليهم الأعراض ويعرفون بإصابتهم بالمرض عرضيًا من خلال فحص دم روتني. من المهم معرفة ما إذا كان المريض قد تعرض سابقًا لعلاج كيماوي أو إشعاعات. تعتبر الحمى ونقص الوزن دليلين على وجود خلل تَكاثُرِيٌّ نِقْوِيّ وليس على خلل التنسج النقوي، يعتبر الأطفال المصابون بمتلازمة داون أكثر عرضة لمتلازمة خلل التنسج النقوي، كما أن تاريخ العائلة يمكن أن يتضمّن مُتَلاَزِمَةُ دي توني-فانكوني (1- نقص تنسج عناصر الدم 2- فرط سيستين الدم) أو فَقْرُ الدَّمِ الوراثي الحَدِيْدِيُّ الأَرُوْمَات.
إن متوسط عمر تشخيص الإنسان بمتلازمة خلل التنسج النقوي 65 عامًا، غير أنه تم تسجيل حالات إصابة بالمرض عند أعمار أقل من ذلك. بعض المرضى قد سبق وتعرضوا لعلاج كيماوي خاصة باستخدام عوامل مؤلكلة مثل: ميفالان، سِيكْلُوفُسْفاميد، بُوسَلْفان وكلورامْبُوسيل. آخرون تعرضوا لإشعاعات (سواء كانت بهدف العلاج أو نتيجة حادثة عرضية) والبعض تعرض لكلا الأمرين (في وقت زراعة الخلايا الجذعية لعلاج مرض آخر على سبيل المثال).يعاني العاملون الذين يتعرضون أكثر من غيرهم للمواد الهيدروكربونية (مثل العمل في قطاع الصناعات البترولية) من صعوبة أكبر من غيرهم في السيطرة على المرض، كما أن التعرض للبنزين وزايلين ارتبط بخلل التنسّج النقوي. ترتفع نسبة الإصابة عند الذكور بفارق بسيط عن الإناث، كما أن المحاربون الفيتناميين الذين تعرضوا للعامل البرتقالي معرضون للإصابة بالمرض. في دراسة صادرة عن الجمعية الأمريكية لدراسات الأورام السريرية في 2010 افترضت وجود علاقة بين تطور متلازمة خلل التنسج النقوي عند الناجين من القنبلة الذرية بعد تعرضهم لتلك الإشعاعات ب 40 إلى 60 عامًا. (في هذه الحالة تم الرجوع إلى الناس الذين كانوا قريبين من مكان انفجار القنبلة الذرية في هيروشيما وناغازاكي خلال الحرب العالمية الثانية).
الخصائص المستخدمة عادةً لتوصيف متلازمة خلل التنسّج النقوي هي: انخفاض تعداد خلايا الدم، تكوّن الدم غير الفعّال، خَلَلُ تَكَوُّنِ الكُرَيَّاتِ الحُمْر، خلل تَكَوُّنُ المُحَبَّبات، وارتفاع تعداد العدلات.
يمكن للثدن أن يؤثر على السلالات الثلاث الموجودة في نخاع العظم. إن أفضل طريقة لتشخيص الثدن هي من خلال فحص شكل الخلايا (morphology) وملوّنات إلكترونية خاصة (PAS) تستخدم في رُشافة نخاع العظم ولُطاخة أو مسحة الدم الطرفي. الثدن في سلسلة النقويات يُعرّف بـ:
- سلسلة المحببات
- العدلات المقطعة بكثرة (تتكرر أيضًا في نقص فيتامين B12 ونقص الفولات (مِلْحُ حَمْضِ الفوليك).
- العدلات قليلة التقطّع (شُذوذُ بيلغِر هُويَه النَّوَوِيّ الكاذب).
- العدلات قليلة الحبيبات أو مُتَلاَزِمَةُ تشيدياق-هيغاشي الكاذبة كبيرة الحبيبات.
- أجسام آور - أوتوماتيكيًا فقر الدم الحرون مع فيض الأرومات II (إذا كان تعداد الأرومات أقل من 5% في الدم الطرفي وأقل من 10% في رُشافة نخاع العظم) أيضًا يمكن رؤية أجسام آور في العدلات الناضجة في مرض ابيضاض الدم النقوي الحاد مع تبادل مواقع (t(8;21.
- الحبيبات ثنائية الشكل (حبيبات قَاعدية ويُوزِيْنِيّة) في الحُبَيباتٌ اليوزِينِيَّة.
- سلسلة الكريات الحمر
- كريات الدم الحمراء الأم ثنائية التنوّي وتَمَزُّقُ النَّواة.
- تبرعم أنوية كريات الدم الحمراء.
- الخيوط النووية لكريات الدم الحمراء أو الجسور بين النووية (تظهر أيضًا في فقر الدم الناتج عن خلل تكوّن كريات الدم الحمراء الخلقي).
- فقدان E-cadherin في الأَرومَةُ الحَمْراءِ السَّوِيَّة هي علامة للشذوذ.
- ملوّن شيف- حمض البيروديك PAS (كروي في الفجوات أو منتشر في التلوين الهيوليّ) ضمن خلايا الدم الحمراء الأم في رُشافة نخاع العظم (ليس له ارتباط بعينة نخاع العظم الثابت البارافينية). ملاحظة: يمكن رؤية نتيجة إيجابية لحمض البيروديك في الفجوات في الأرومات من نوع L1 و L2. (هذه التسمية مستخدمة في النظام الأمريكي الفرنسي البريطاني)
- الأرومات الحديدية الحلقية تظهر في لطخة حديد زُرْقَةُ بروسيا (10 حبيبات من الحديد أو أكثر في 1/3 النواة أو أكثر، عندما أجري تعداد على خلايا الدم الحمراء الأم كانت أكثر من 15% منها عبارة عن أرومات حديدية حلقية).
- سلسلة النّوّاءات (قد تكون الأكثر شخصانية-غير موضوعي)
- الصفائح ذات الأنوية قليلة التقطّع ينتج عنها نوّاءات (نقص في التَفَصُّص).
- النوّاءات كثيرة التقطع.
- انتفاخ الصفائح (يُشاهد من خلال التداخل باستخدام المجهر).
اللطخات الأخرى يمكن أن تساعد في حالات خاصة (ملوّن شيف- حمض البيروديك والنافثول) تستخدم في اليوزينِيَّات كعلامة الخلل الظاهر في فقر الدم اليوزينِيَّ المزمن والحالة الشاذة.
عند فحص نخاع العظم؛ الدرجة العالية من الثدن (فقر الدم الحرون مع فيض الأرومات I و II) قد يظهر تموضع غير اعتيادي للخلايا الأم غير الناضجة، وهي عبارة عن جزر من الخلايا الأم غير الناضجة (الأرومات النقوية و promyelcytes) التي تتموضع في وسط الفراغ البين تَرْبيقِيّ بدلًا من أن يكون محاذيًا للترابيق أو محيطًا بالشرايين. شكل الخلايا هذا قد يصعب تمييزه عن فقر الدم المعالج ووعناصر نخاع العظم غير الناضجة المتماثلة للشفاء. كما أن التغيرات الطبوغرافية لكريات الدم الحمراء ذات الأنوية يمكن أن تُرى في الحالات المبكرة من خلل التنسج النقوي (فقر الدم الحرون وفقر الدم الحرون مع الأرومات الحديدية الحلقية)؛ حيث تجد الأرومات السوية إلى جانب التربيق العظمي بدلًا من تشكيل جزر من خلايا الدم الحمراء في الفراغات بي الخلوية بشكل طبيعي.
خلل التنسّج النقوي هو تشخيص يعتمد على الاستثناء ولا يتّخذ قرار بشأنه إلّا بعد تحديد دقيق لوضع مخازن الحديد، نقص الفيتامينات ونقص التغذية.
العلاج
إن أهداف العلاج تتمثل في التحكم في الأعراض، تحسين نوعية حياة المريض، زيادة احتمالية النجاة وتخفيض تقدّم مرض ابيضاض الدم النقوي الحاد.
النظام العالمي لقياس أعراض البروستاتا IPSS.[15] يمكن أن يساعد المرضى المحجورين صحيًا لعلاج أكثر حدة (بمعنى آخر: زراعة نخاع العظم) كما أنه يساعد في تحديد أفضل توقيت للقيام بهذا العلاج.[16] أركان هذا العلاج الرئيسية تتمثل في العناية المساعدة إلى جانب دعم منتجات الدم المساعدة وعوامل النمو المتعلقة بالدم مثل إريثروبويَتين. القوانين المنظمة لاستخدام إريثروبويَتين في طور التطور حاليًا وفق الجمعية الأمريكية العالمية للعناية الطبية. لم يرد شيء بخصوص استخدام عوامل النمو المتعلقة بالدم لعلاج متلازمة خلل التنسج النقوي في تلك الوثيقة.[17]
وافقت منظمة الغذاء والدواء الأمريكية على استخدام ثلاثة عوامل في علاج متلازمة خلل التنسّج النقوي:
- 5-آزاسيتيدين: متوسط النجاة 21 شهرًا.[18][19][20][21]
- ديسيتابين: معدل الاستجابة الكاملة وصل لـ 43%. أظهرت دراسة أنه إذا استخدم مع حمض الفالبوريك يظهر فاعلية في علاج ابيضاض الدم النقوي الحاد.[22][23][24][25]
- ليناليدوميد: يظهر فاعلية في تخفيض حاجة مرضى متلازمة 5q لنقل كريات الدم الحمراء لهم.[26]
العلاج الكيماوي مع العوامل دون المثيلية، 5- آزاسيتيدين وديسيتابين أظهر أنه يقلل الحاجة لنقل الدم كما أنه يعيق تطور متلازمة خلل التنسج النقوي إلى ابيضاض الدم النقوي الحاد. في ديسيمبر 2005 تمت موافقة منظمة الغذاء والدواء الأمريكية على عقار ليناليوميد ليتم استخدامه في حالات متلازمة 5q فقط. علاج متلازمة خلل النسج النقوي باستخدام ليناليدوميد في الولايات المتحدة يكلّف (9200 $) شهريًا.[27]
زراعة الخلايا الجذعية، تحديدًا عند المرضى صغار السن (أقل من 40 سنة) والمرضى الأكثر تضررًا؛ تشكّل العلاج الشافي. يرتبط نجاح عملية زراعة نخاع العظم بشدة متلازمة خلل التنسّج النقوي وفقًا للنظام العالمي لقياس أعراض البروستاتا IPSS، المرضى الذين يمتلكون نتائج أكثر تفضيلًا وفقًا لهذا النظام يحصلون على نتائج أفضل لزراعة نخاع العظم.[28]
تعتبر زيادة عنصر الحديد ونقل كريات الدم الحمراء جزءًا رئيسيًا من العلاج المساعد لمرضى متلازمة خلل التنسج النقوي الذين يعانون من فقر الدم الأنيمي. مع أن بعض العلاجات التي يأخذها المرضى يمكن أن تزيد الحاجة إلى نقل كريات الدم الحمراء للمريض إلا أن بعض المرضى قد لا يستجيبون لهذه العلاجات وقد يعانون بمرور الوقت من تراكم الحديد وما ينتج عن ارتفاع نسبته من مضاعفات.
المرضى الذين يحتاجون إلى نقل كريات الدم الحمراء بأعداد كبيرة، يعانون من آثار عكسية تتمثل في ارتفاع كميات الحديد في كل من الكبد، القلب وجهاز الغدد الصم. من المحتمل أن يكون الخلل في عمل هذه الأعضاء ناتجًا عن ارتفاع كميات الحديد فيها عند مرضى متلازمة خلل التنسج النقوي، كما أن ارتفاع الحديد الناتج عن نقل الدم قد يكون مساهمًا في زيادة الأمراض والوفيات في المراحل المبكّرة من خلل التنسّج النقوي.
عند المرضى الذين يحتاجون نقل كريات الدم الحمراء لهم، يجب مراقبة مستوى الفريتين في مصل الدم، عدد كريات الدم الحمراء المنقولة وتعطّل عمل بعض الأعضاء (القلب، الكبد، البنكرياس) لمعرفة مستوى الحديد. يجب أن يُخفّض مستوى الحديد إلى أقل من 1000 مكغ/لتر. يتوافر الآن في الولايات المتحدة خالبان للحديد؛ دِيفِيرُوكْسامِين للاستخدام بشكل وريدي، وديفيراسيروكس للاستخدام الفموي. هذه الخيارات توفّر الآن علاجًا ناجعًا محتملًا لمشكلة اكتناز الحديد، ديفيريبرون هو عامل خالب ثالث يتوافر في أوروبا للاستخدام الفموي ولا يتوافر في الولايات المتحدة. الدراسات السريرية الخاصة بمتلازمة خلل التنسج النقوي ما زالت قائمة مع عوامل الحديد الخالبة لمعرفة ما إذا كالنت خوالب الحديد ستغيّر من طبيعة المرضى الذين يحتاجون لنقل الدم. لقد أمكن التغلب على بعض مضاعفات اكتناز الحديد عند مرضى متلازمة نقص التنسج النقوي من خلال استخدام علاج مخالب الحديد.
حاليًا؛ هناك عدد هائل من التجارب السريرية التي تجري على الطور الثالث مقارنةً بالعلاج باستخدام ديفيراسيروكس الذي يجب أن يساعد في تحديد فائدته في علاج اكتناز الحديد عند مرضى متلازمة خلل التنسج النقوي.
في 2013 في ألمانيا أجريت دراسة على الطور الأول مع APG101 عند المرضى الذين يعتمدون على نقل الدم لهم مع احتمالات قليلة أو متوسطة بأن يكونوا بخطر الإصابة بمتلازمة خلل التنسج النقوي. التجارب قبل السريرية أظهرت أن APG101 قد أنقذت خلايا الدم الحمراء الأم من الموت في مرحلة ما قبل النضج. لذا المعالجة باستخدام APG101 من المتوقع أن تُحسّن تَكَوُّنُ الكُرَيَّاتِ الحُمْر وفقر الدم عند مرضى متلازمة خلل التنسّج النقوي. الهدف الرئيسي من الدراسة هو اختبار سلامة استخدام APG101 واستجابة خلايا الدم عند مرضى متلازمة خلل التنسج.[29]
توقعات سير المرض
التوقعات المستقبلية بشأن مرضى متلازمة خلل التنسّج النقوي مختلفة؛ 30% من المرضى تتطور حالتهم إلى ابيضاض الدم النقوي الحاد. متوسط معدلات النجاة تتراوح بين سنوات إلى أشهر حسب نوع المرض. زراعة الخلايا الجذعية توفر علاجًا للمرض مع معدلات نجاة قد تصل إلى 50% من الحالات خلال 3 سنوات ، وتنخفض احتمالات النجاة مع تقدم العمر.[30]
دلائل المآل الجيد للمرض: الأعمار الصغيرة؛ نقصان عادي أو متوسط في تعداد العدلات أو الصفائح، نقصان في تعداد الأرومات في نخاع العظم إلى ما دون 20% وانعدام وجود الأرومات في الدم، انعدام وجود أجسام آور، وجود أرومات حديدية حلقية، أنماط نووية عادية لأنماط نووية مركبة بلا وجود اختلالات كروموسومية، وفي المختبر؛ زراعة النخاع تظهر نموًا.
دلائل المآل السيء للمرض: الأعمار المتقدمة؛ انخفاض شديد في تعداد العدلات أو كريات الدم الحمراء، ارتفاع تعداد الأرومات في نخاع العظم (20-29%) أو في الدم، أجسام آور، غياب الأرومات الحديدية الحلقية، تموضع غير طبيعي أو عدم نضج للخلايا الأم المحببة في نخاع العظم أو أنماط نووية غير اعتيادية أو اعتلالات في كروموسومات النخاع، وفي المختبر؛ زراعة النخاع أظهرت نموًا يشير لابيضاض الدم . سير المرض والأنماط النووية: جيد: طبيعي، -Y، del(5q)، del(20q) متوسط أو متغيّر: +8، شذوذات أخرى مفردة أو على شكل أزواج. ضعيف: مركب (أقل من 3 انحرافات صبغية)، شذوذ أو انحراف في كروموسوم 7.[31]
النظام العالمي لقياس أعراض البروستاتا IPSS هي أكثر أداة مستخدمة في توقع سير متلازمة خلل التنسج النقوي.[32]
الاعتلالات في خريطة الصبغيات يمكن تحديدها باستخدام خريطة صبغيات طبيعية، فحص التهجين الموضعي المشع FISH أو الأنماط النووية الافتراضية.
الوبائيات
إن عدد المصابين بمتلازمة خلل التنسج النقوي غير معروف على وجه التحديد؛ لأن كثيرًا من الحالات لم يتم تشخيصها بالإضافة إلى انه لم يكن هناك تكليف بتتبع المتلازمة . تتحدث بعض التخمينات عن زيادة 10000 إلى 20000 حالة من هذا المرض في الولايات المتحد الأمريكية وحدها سنويًا. من المحتمل أن تكون زيادة الوقوعات مصاحبة لزيادة عمر النسمة، ويتوقع البعض أن الوقوعات عند المرضى فوق 70 سنة قد تزيد بمعدّل 15 حالة لكل 100000 سنويًا.[33]
تاريخ المرض
منذ بدايات القرن العشرين؛ بدأ تمييز بعض الناس الذين يعانون من ابيضاض الدم النقوي الحاد أنهم سبق وعايشوا فترة من فقر الدم وانتاج خلايا الدم غير العاديّ، لقد جُمعت هذه الحالات مع أمراض أخرى تحت مسمى "فقر الدم الحرون". أول توصيف لـ "مُقَدِّماتُ الاَبْيِضاض" ككينونة محددة تم نشره عام 1953 بواسطة بلوك جاكبسون.[34] لقد كانت بدايات التعرف، التمييز والتصنيف لهذا المرض مثيرة للجدل، وتغيرت تسميات المتلازمة حتى عام 1976 عندما نُشر التصنيف الفرنسي الأمريكي البريطاني وانتشر اسمها الحالي؛ متلازمة خلل التنسّج النقوي.
مرضى مشهورون
تتضمن قائمة الأشخاص المشهورين الذين أصيبوا بمرض متلازمة خلل التنسج:
- عالم الفلك كارل ساجان.
- مدرّب التجديف في جامعة هارفرد هاري باركر.[35]
- الفائز بجائزة نوبل لفرع الكيمياء ألان ماكديرميد.
- الكاتبة سوزان سونتاج.
- الكاتبة نورا إيفرون.
- الكاتب روالد دال.
- المصورة الفوتوغرافية ماري إلين مارك.
- موسيقيي الجاز مايكل بريكر، جوي فاريل وباول موتيان.
- الممثلة نينا فوخ.
- مُنفّذ الألعاب النارية فال فورجيت.
- عضوي الكونغرس الأمريكي جوي موآكلي وبوب ماتسوي.
- الممثل بات هينغل.
- الممثل لاري هاغمان.
- الممثلة والكوميدية فران أليسون.
- شخصية الراديو جي. بي. ماكارثي
- عالم الفيزياء النظرية الدنيماركي بير باك.
- أول فائز في مسابقة نحلة التهجئة القومية فرانك نوهاسور.
- مضيف برنامج صباح الخير أمريكا روبن روبرتس.
- الممثل أمريش بوري.
- الرسّام بوني لي باتلير.
- الشاعر جون غليمان.
- لاعب الهوكي المحترف دوج موهنس.
- الممثل/ الكوميدي ديف مادّين.
- الناشط الطبي لويل موس.
- مصمم الأزياء كريستيان أودياجير.
المراجع
- منظمة الصحة العالمية، ميزة نمو خلايا CD34+ في التثليث الصبغي 8 لمتلازمة خلل التنسج النخاعي العالية الخطورة بالرغم من تحسن إشارات الاستماتة نسخة محفوظة 05 نوفمبر 2018 على موقع واي باك مشين.
- "myelodysplastic syndrome" في معجم دورلاند الطبي
- page 393 First Aid USMLE 2014, Author Tao Le, McGraw Hill Education
- Bennett JM, Catovsky D, Daniel MT, et al. (August 1976). "Proposals for the classification of the acute leukaemias. French-American-British (FAB) co-operative group". Br. J. Haematol. 33 (4): 451–8. doi:10.1111/j.1365-2141.1976.tb03563.x. PMID 188440. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Table 1: French-American-British (FAB) Classification of MDS". مؤرشف من الأصل في 7 أكتوبر 2008. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Greenberg P, Cox C, LeBeau MM, Fenaux P, Morel P, Sanz G, Sanz M, Vallespi T, Hamblin T, Oscier D, Ohyashiki K, Toyama K, Aul C, Mufti G, Bennett J (1997). "International scoring system for evaluating prognosis in myelodysplastic syndromes". Blood. 89 (6): 2079–88. PMID 9058730. الوسيط
|CitationClass=تم تجاهله (مساعدة)صيانة CS1: أسماء متعددة: قائمة المؤلفون (link) - Myelodysplastic Syndrome. The Leukemia & Lymphoma Society. White Plains, NY. 2001. p 24. Retrieved 05-12-2008.
- Cazzola M, Invernizzi R, Bergamaschi G, et al. (2003). "Mitochondrial ferritin expression in erythroid cells from patients with sideroblastic anemia". Blood. 101 (5): 1996–2000. doi:10.1182/blood-2002-07-2006. PMID 12406866. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Bunn HF (1986). "5q- and disordered haematopoiesis". Clinics in haematology. 15 (4): 1023–35. PMID 3552346. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Van den Berghe H, Cassiman JJ, David G, Fryns JP, Michaux JL, Sokal G (1974). "Distinct haematological disorder with deletion of long arm of no. 5 chromosome". Nature. 251 (5474): 437–8. doi:10.1038/251437a0. PMID 4421285. الوسيط
|CitationClass=تم تجاهله (مساعدة)صيانة CS1: أسماء متعددة: قائمة المؤلفون (link) - List A, Kurtin S, Roe DJ, et al. (2005). "Efficacy of lenalidomide in myelodysplastic syndromes". N. Engl. J. Med. 352 (6): 549–57. doi:10.1056/NEJMoa041668. PMID 15703420. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Rozovski U, Keating M, Estrov Z (2012) The significance of spliceosome mutations in chronic lymphocytic leukemia. Leuk Lymphoma
- Gondek LP, Tiu R, O'Keefe CL, Sekeres MA, Theil KS, Maciejewski JP. Chromosomal lesions and uniparental disomy detected by SNP arrays in MDS, MDS/MPD, and MDS-derived AML. Blood. 2008 Feb 1;111(3):1534-42.
- Copper deficiency causes reversible myelodysplasia - Huff - 2007 - American Journal of Hematology - Wiley Online Library نسخة محفوظة 29 ديسمبر 2016 على موقع واي باك مشين.
- MDS - Myelodysplastic Syndromes نسخة محفوظة 09 أبريل 2017 على موقع واي باك مشين.
- Cutler CS, Lee SJ, Greenberg P, Deeg HJ, Perez WS, Anasetti C, Bolwell BJ, Cairo MS, Gale RP, Klein JP, Lazarus HM, Liesveld JL, McCarthy PL, Milone GA, Rizzo JD, Schultz KR, Trigg ME, Keating A, Weisdorf DJ, Antin JH, Horowitz MM (2004). "A decision analysis of allogeneic bone marrow transplantation for the myelodysplastic syndromes: delayed transplantation for low-risk myelodysplasia is associated with improved outcome". Blood. 104 (2): 579–85. doi:10.1182/blood-2004-01-0338. PMID 15039286. الوسيط
|CitationClass=تم تجاهله (مساعدة)صيانة CS1: أسماء متعددة: قائمة المؤلفون (link) - "Centers for Medicare & Medicaid Services". مؤرشف من الأصل في 14 يناير 2009. اطلع عليه بتاريخ 29 أكتوبر 2007. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Wijermans P, Lübbert M, Verhoef G, et al. (2000). "Low-dose 5-aza-2'-deoxycytidine, a DNA hypomethylating agent, for the treatment of high-risk myelodysplastic syndrome: a multicenter phase II study in elderly patients". J. Clin. Oncol. 18 (5): 956–62. PMID 10694544. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Lübbert M, Wijermans P, Kunzmann R, et al. (2001). "Cytogenetic responses in high-risk myelodysplastic syndrome following low-dose treatment with the DNA methylation inhibitor 5-aza-2'-deoxycytidine". Br. J. Haematol. 114 (2): 349–57. doi:10.1046/j.1365-2141.2001.02933.x. PMID 11529854. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Silverman LR, Demakos EP, Peterson BL, et al. (2002). "Randomized controlled trial of azacitidine in patients with the myelodysplastic syndrome: a study of the cancer and leukemia group B". J. Clin. Oncol. 20 (10): 2429–40. doi:10.1200/JCO.2002.04.117. PMID 12011120. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Silverman LR, McKenzie DR, Peterson BL, et al. (2006). "Further analysis of trials with azacitidine in patients with myelodysplastic syndrome: studies 8421, 8921, and 9221 by the Cancer and Leukemia Group B". J. Clin. Oncol. 24 (24): 3895–903. doi:10.1200/JCO.2005.05.4346. PMID 16921040. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Kantarjian HM, O'Brien S, Shan J, et al. (2007). "Update of the decitabine experience in higher risk myelodysplastic syndrome and analysis of prognostic factors associated with outcome". Cancer. 109 (2): 265–73. doi:10.1002/cncr.22376. PMID 17133405. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Kantarjian H, Issa JP, Rosenfeld CS, et al. (2006). "Decitabine improves patient outcomes in myelodysplastic syndromes: results of a phase III randomized study". Cancer. 106 (8): 1794–803. doi:10.1002/cncr.21792. PMID 16532500. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Kantarjian H, Oki Y, Garcia-Manero G, et al. (2007). "Results of a randomized study of 3 schedules of low-dose decitabine in higher-risk myelodysplastic syndrome and chronic myelomonocytic leukemia". Blood. 109 (1): 52–7. doi:10.1182/blood-2006-05-021162. PMID 16882708. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Blum W, Klisovic RB, Hackanson B, et al. (2007). "Phase I study of decitabine alone or in combination with valproic acid in acute myeloid leukemia". J. Clin. Oncol. 25 (25): 3884–91. doi:10.1200/JCO.2006.09.4169. PMID 17679729. الوسيط
|CitationClass=تم تجاهله (مساعدة) - List A, Dewald G, Bennett J, et al. (2006). "Lenalidomide in the myelodysplastic syndrome with chromosome 5q deletion". N. Engl. J. Med. 355 (14): 1456–65. doi:10.1056/NEJMoa061292. PMID 17021321. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Lenalidomide (Revlimid) for anemia of myelodysplastic syndrome". The Medical letter on drugs and therapeutics. 48 (1232): 31–2. 2006. PMID 16625140. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Oosterveld M, Wittebol S, Lemmens W, Kiemeney B, Catik A, Muus P, Schattenberg A, de Witte T (2003). "The impact of intensive antileukaemic treatment strategies on prognosis of myelodysplastic syndrome patients aged less than 61 years according to International Prognostic Scoring System risk groups". Br J Haematol. 123 (1): 81–9. doi:10.1046/j.1365-2141.2003.04544.x. PMID 14510946. الوسيط
|CitationClass=تم تجاهله (مساعدة)صيانة CS1: أسماء متعددة: قائمة المؤلفون (link) - Home - ClinicalTrials.gov نسخة محفوظة 04 يوليو 2018 على موقع واي باك مشين.
- Kasper, Dennis L; Braunwald, Eugene; Fauci, Anthony; et al. (2005). Harrison's Principles of Internal Medicine (الطبعة 16th). New York: McGraw-Hill. صفحة 625. ISBN 0-07-139140-1. الوسيط
|CitationClass=تم تجاهله (مساعدة)صيانة CS1: أسماء متعددة: قائمة المؤلفون (link) - Solé E, et al. (2000). "Incidence, characterization and prognostic significance of chromosomal abnormalities in 640 patients with primary myelodysplastic syndromes". British Journal of Haematology. 108 (2): 346–356. doi:10.1046/j.1365-2141.2000.01868.x. PMID 10691865. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Greenberg; et al. (1997). "International Scoring System for Evaluating Prognosis in Myelodysplastic Syndromes". Blood. 89: 2079–2088. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Aul C, Giagounidis A, Germing U (2001). "Epidemiological features of myelodysplastic syndromes: results from regional cancer surveys and hospital-based statistics". Int. J. Hematol. 73 (4): 405–10. doi:10.1007/BF02994001. PMID 11503953. الوسيط
|CitationClass=تم تجاهله (مساعدة)صيانة CS1: أسماء متعددة: قائمة المؤلفون (link) - Block M, Jacobson LO, Bethard WF (July 1953). "Preleukemic acute human leukemia". J Am Med Assoc. 152 (11): 1018–28. doi:10.1001/jama.1953.03690110032010. PMID 13052490. الوسيط
|CitationClass=تم تجاهله (مساعدة)صيانة CS1: أسماء متعددة: قائمة المؤلفون (link) - Hewitt, Ed (25 June 2013). "Harry Parker has passed; a remembrance, 1935-2013". Row2k. مؤرشف من الأصل في 2 أكتوبر 2017. اطلع عليه بتاريخ 27 يونيو 2013. الوسيط
|CitationClass=تم تجاهله (مساعدة)
 بوابة طب
بوابة طب
