هيموفيليا
نزف الدم الوراثي[1] أو الهيموفيليا[2] أو الناعور[2] أو النزاف[2] (بالإنجليزية: haemophilia) هو الاسم الذي يٌطلق على أي من الأمراض الوراثية المتعددة التي تسبب خللا في الجسم وتمنعه من السيطرة على عملية تخثر الدم. إن الأسباب الوراثية (أو نادرًا، أسباب في المناعة الذاتية للجسم) تسبب نقصا في عوامل تخثر البلازما الذي يعمل على تسوية عملية تخثر الدم، عندما يصاب وعاء دموي بجرح لن تتكون خثرة ويستمر الدم بالتدفق عوامل التخثر لمدة طويلة من الزمن. يمكن للنزيف أن يكون خارجيًا، كالجلد إذا تم حكه بشيء أو عندما يُصاب بقطع، أو أن يكون النزيف ظاهراً أي في الكدمات التي على الجلد، نزيفا داخلياً كنزيف الأمعاء أو النزيف الدماغي أو النزيف في العضلات أو المفاصل أو الأعضاء المجوفة.
| الناعور نزف الدم الوراثي النزاف الهيموفيليا | |
|---|---|
 يعد نقص العامل الثامن أكثر الأسباب شيوعا. يعد نقص العامل الثامن أكثر الأسباب شيوعا. | |
| النطق | /hiːməˈfɪliə/ |
| معلومات عامة | |
| الاختصاص | علم الدم |
| التاريخ | |
| وصفها المصدر | قاموس بروكهاوس وإفرون الموسوعي |
ومثل الاضطرابات المتنحية الأخرى المرتبطة بالجنس، اضطرابات الصبغي (إكس)، يحدث الناعور في الذكور أكثر من الإناث. وذلك لأن الإناث يحملن اثنين من الصبغيات إكس بينما الذكور واحد فقط، لذلك فظهور المرض مضمون في أي من الذكور الذين يحملونه. فرصة وجود نسختين من الجينات المعيبة في الإناث بعيدة جدًا، لذلك الإناث غالبًا حاملات للمرض وينقلنه بدون أن تظهر عليهن أعراض. ترث الإناث الجينات المعيبة من الأم أو الأب أو قد تكون طفرة جديدة، على الرغم من ذلك؛ ليس مستحيلًا إصابة أنثى بمرض سيولة الدم.
التشخيص
التصنيف
هناك عدة أنواع من الهيموفيليا: الهيموفيليا أ، والهيموفيليا ب، والهيموفيليا ج، والباراهيموفيليا، والهيموفيليا أ المكتسبة، والهيموفيليا ب المكتسبة.[3][4][5][6]
الهيموفيليا أ، هي اضطراب وراثي متنحٍّ مرتبط بالصبغي إكس يؤدي إلى نقص عامل التخثر الوظيفي الثامن. الهيموفيليا ب، هي أيضًا اضطراب وراثي متنحٍّ مرتبط بالصبغي إكس يتضمن نقص عامل التخثر الوظيفي التاسع. الهيموفيليا ج، هي اضطراب وراثي جسمي ينطوي على نقص عامل التخثر الوظيفي الحادي عشر. الهيموفيليا ج ليست اضطرابًا متنحيًا تمامًا، لأن الأفراد غير المتجانسين بالصفات الوراثية يُظهرون أيضًا نزيفًا متزايدًا.[7]
نوع الهيموفيليا المعروف باسم باراهيموفيليا هو شكل خفيف ونادر سببه نقص في العامل الخامس. يمكن أن يكون هذا النوع موروثًا أو مكتسبًا.
يحدث الشكل غير الوراثي للهيموفيليا الناجم عن الأضداد الذاتية للعامل الثامن، ويُعرف أيضًا باسم الهيموفيليا أ المكتسبة. يمكن أن ترتبط الهيموفيليا المكتسبة بالسرطانات، واضطرابات المناعة الذاتية، وما بعد الولادة.[8]
التاريخ
المعالجة
لا يوجد علاج طويل الأمد. إن معالجة نوبات النزف والوقاية منها تجريان بشكل أساسي بواسطة تعويض عوامل تخثر الدم المفقودة.[9]
الإدارة
عوامل التخثر
ليس هناك حاجة لعوامل التخثر في حالات الهيموفيليا الخفيفة عادةً. في حالات الهيموفيليا متوسطة الشدة، ليس هناك حاجة لعوامل التخثر عادةً إلا عند حدوث النزيف أو من أجل الوقاية منه في ظروف معينة. في حالات الهيموفيليا الشديدة، يوصى بالرعاية الوقائية مرتين أو ثلاث مرات في الأسبوع، ويمكن أن تستمر مدى الحياة. يؤدي العلاج السريع لحوادث النزيف إلى تقليل الضرر الذي يلحق بالجسم.[10]
يُستخدم العامل الثامن في الهيموفيليا أ والعامل التاسع في الهيموفيليا ب. يمكن التعويض إما بواسطة العامل المعزول من مصل الدم البشري، أو العامل المعاد تركيبه (المأشوب)، أو بواسطة مزيج من الاثنين. يطور بعض الأشخاص أجسامًا مضادة (مثبطات) ضد العوامل التعويضية المعطاة لهم، لذلك يجب زيادة مقدار العامل أو إعطاء منتجات بديلة غير بشرية، مثل العامل الثامن المأخوذ من الخنزير.
إذا أصبح الشخص مقاومًا للعلاج عبر تعويض عامل التخثر نتيجةً لمستويات عالية من المثبطات الجائلة في الدوران، فيمكن التغلّب على هذا الأمر جزئيًا باستخدام العامل البشري الثامن المُعاد تركيبه.
في أوائل عام 2008، وافقت إدارة الغذاء والدواء الأمريكية (إف دي إيه) على عامل مضاد للهيموفيليا رُكّب وراثيًا من جينات خلايا بويضة الهامستر الصيني. منذ عام 1993، كانت منتجات العوامل المعاد تركيبها (التي تُصَنّع عادةً من خلايا الأنسجة المُستنبتة لبويضة الهامستر الصيني، والتي تتضمن القليل من منتجات بلازما الدم البشرية) متاحة ومستخدمة على نطاق واسع في الدول الغربية الأكثر ثراءً. في حين أن منتجات عامل التخثر المعاد تركيبه توفر درجة أعلى من النقاء والسلامة، فإنها، باعتبارها مركّزة، مكلفةٌ للغاية، وغير متاحة عمومًا في العالم النامي. في كثير من الحالات، يصعب الحصول على منتجات العوامل من أي نوع في البلدان النامية.
تُعطى عوامل التخثر إما بشكل وقائي أو عند الحاجة. يتضمن الاستخدام الوقائي ضخ عامل التخثر وفقًا لجدول زمني منتظم للحفاظ على مستويات التخثر مرتفعة بما يكفي لمنع نوبات النزيف التلقائي. يتضمن العلاج عند الحاجة (أو العرضي) علاج نوبات النزف بمجرد ظهورها. في عام 2007، قارنت تجربة بين العلاج عند الحاجة للأولاد (أقل من 30 شهرًا) المصابين بالهيموفيليا أ مع العلاج الوقائي (إعطاء 25 وحدة دولية لكل كيلوغرام من وزن الجسم من العامل الثامن كل يوم) من ناحية التأثير على منع حدوث المرض المفصلي. عندما بلغ الأولاد 6 سنوات، كان 93% من أفراد مجموعة الوقاية و55% من أفراد مجموعة العلاج العَرَضي لديهم بنية مفصلية طبيعية عند التصوير بالرنين المغناطيسي. ومع ذلك، نتج عن العلاج الوقائي متوسط تكاليف قدرها 300,000 دولار في السنة. يؤيد صاحب المقال الافتتاحي الذي نُشر في نفس العدد من مجلة نيو إنغلاند جورنال أوف ميديسين فكرة أن العلاج الوقائي ليس أكثر فعالية من العلاج عند الحاجة فقط، بل يشير أيضًا إلى أن البدء بعد أول نزيف خطير مرتبط بالمفاصل يمكن أن يكون أكثر فعالية من ناحية التكلفة من الانتظار حتى بلوغ السن الثابتة للبدء. لدى معظم مرضى الهيموفيليا في بلدان العالم الثالث وصول محدود أو معدوم إلى منتجات عوامل تخثر الدم التجارية.[11][12][13]
العلامات والأعراض
الأعراض المميزة تختلف مع شدة المرض. بشكل عام الأعراض عبارة عن نوبات من النزيف الداخلي أو الخارجي. تتراوح شدة النزف بين المعتدل أو الخطير ولكن حتى في الهيموفيليا المعتدلة تزداد الأعراض بعد الجراحة أو الصدمات الخطيرة. في كل من الناعور أ وب، هناك نزيف تلقائي ولكن زمن فترة النزيف طبيعي، وقت البروثرومبين طبيعي، وقت الثرومبين طبيعي، ولكن يطول وقت تجلط الدم الجزئي. النزيف الداخلي شائع في الناس مع الهيموفيليا الشديدة وبعض الأفراد الذين يعانون من الهيموفيليا المعتدلة. النوع الأكثر تميزًا للنزف الداخلي هو نزف المفاصل [14] وحتى يمكن أن يحدث من تلقاء نفسه (من دون صدمة واضحة). إذا لم يعالج بسرعة، يمكن أن يؤدي النزيف إلى تلف المفاصل الدائم والتشوه.[14] النزيف في الأنسجة الرخوة مثل العضلات والأنسجة تحت الجلد أقل حدة ولكن يمكن أن يؤدي إلى تلف لذا يتطلب العلاج.
الأطفال الذين يعانون من الناعور المعتدل قد لا تظهر أي علامات أو أعراض لديهم عند الولادة خاصةً إذا لم تحدث لهم عملية الختان. ظهور أول الأعراض غالبًا ما تكون كدمات وتورمات من السقطات المتكررة أثناء تعلمهم المشي. قد يحدث أيضًا تورم وكدمات من نزيف في المفاصل والأنسجة الرخوة أو العضلات. أو قد تكون أول العلامات هي نزيف حاد من عمليات الأسنان، وقوع حادث، أو الجراحة. الإناث الحاملات للمرض عادة ما يكون لديهن ما يكفي من عوامل التخثُّر لمنع حدوث مشاكل النزيف الخطيرة، ولكن البعض قد يتعرضن للهيموفيليا الخفيفة.
المضاعفات
المضاعفات الخطيرة أكثر شيوعًا في المصابين بنزف الدم الحاد والمعتدل. قد تكون المضاعفات سواء بصورة مباشرة من المرض أو أثناء العلاج:[15]
- نزيف داخلي عميق، على سبيل المثال، نزيف العضلات العميقة، مما يؤدي إلى تورم، وخدر أو آلام الأطراف.
- تلف المفاصل من تدمي المفصل مع الألم الشديد والتشوه، نتيجة لالتهاب المفصل وتدميره.
- الإصابة بالعدوى التي تنتقل من عمليات نقل الدم أثناء العلاج.
- ردود الفعل السلبية أثناء المعالجة بعوامل التخثُّر، بما في ذلك تطوير مثبطات المناعة مما يجعل استبدال العوامل أقل فعالية.
- نزيف بالمخ وهي حالة طبية طارئة ناجمة عن تراكم الضغط داخل الجمجمة. ويمكن أن يسبب الغثيان وفقدان الوعي، تلف المخ، والموت.
يأتي تدمّي المفاصل بسبب الناعور في شكل التهاب الأغشية المفصلية المزمن وتدمير الغضاريف,[16] إذا لم يُسحب الدم من داخل المفصل بسرعة يمكن أن يؤدي إلى موت الخلايا الغضروفية ويؤثر على تركيب البروتيوغليكان. قد تكون بطانة المفاصل المتضخمة والهشة عرضة للنزف مجددًا أثناء محاولة التخلص من الدم المتزايد، مما يؤدي إلى حلقة مفرغة من تدمي المفصل. وبالإضافة إلى ذلك، ترسب الحديد في الغشاء الزليلي قد يحفز استجابة التهابية وتنشيط نظام المناعة، وتحفيز الأوعية الدموية، مما يؤدي لتدمير الغضاريف والعظام.[17]
متوسط العمر المُتوقَّع
مثل معظم جوانب المرض، يتفاوت العمر المتوقع باختلاف شدة المرض وتوافر العلاج المناسب. الأشخاص الذين يعانون من الهيموفيليا الشديدة أو لا يحصلون على علاج مناسب يقل متوسط أعمارهم المتوقعة بشكل كبير، غالبًا لايصلون إلى مرحلة النضج. قبل الستينات عندما أصبح العلاج الفعّال متاحًا، كان متوسط العمر المتوقع 11 عام.[14] وفي الثمانينات كان العمر المتوقع لمرضى الناعور 50-60 سنة. أمااليوم مع توافر العلاج المناسب، فإن الذكور المصابين بالناعور لديهم متوسط أعمار طبيعي، أو ما يقرب من 10 أعوام أقصر من الذكور الأصحاء.[18]
منذ الثمانينات، تحول السبب الرئيسي لوفاة الأشخاص الذين يعانون من الناعور الشديد من النزيف إلى الإصابة بمرض نقص المناعة المكتسبة (الإيدز) أثناء العلاج أو استخدام منتجات الدم الملوثة.[14] ثاني الأسباب الرئيسية للوفاة متعلق بالمضاعفات الخطيرة وهي نزيف المخ، الذي يمثل اليوم ثلث مجموع الوفيات من الناس المصابين بالناعور. تشمل اثنين من الأسباب الرئيسية الأخرى للوفاة التهابات الكبد مسببة التليف وانسداد مجرى الهواء أو تدفق الدم بسبب نزيف الأنسجة الرخوة.[14]
أنواع مرض نزف الدم
- هيموفيليا أ– قلة العامل الثامن وتمثل 80% من حالات سيولة الدم.
- هيموفيليا ب– قلة العامل التاسع وتمثل تقريبًا 20% من الحالات.[19]
- هيموفيليا ج – قلة العامل الحادي عشر وهي اضطراب وراثي أي لا يرتبط بالصبغي إكس.
وتعتبر الهيموفيليا (أ) و(ب) الأكثر انتشاراً في الوطن العربي نتيجة لنقص بروتينات التجلط (8) و(9) على التوالي وتظهر الهيموفيليا (أ) و(ب) بين الذكور دون الإناث ويكون انتقال العامل الوراثي من الأم إلى الابن الذكر وليس من الأب إلى الأبن، حيث أن الابنة هي التي تكون حاملة للمرض وتورثه لأبنائها الذكور دون أن تظهر عليها الأعراض.[20]
جينات
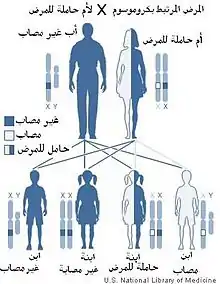
تمتلك الإناث صبغيين X بينما لدى الذكور صبغي Y وآخر X. وبما أن الطفرة المسببة مرتبطة بالصبغي X، تحمل الأنثى المرض على أحد الصبغيين X ولا تكون مٌتأثرة به لأن الصبغي الآخر الذي هو X أيضاً سيعمل على توليد عوامل التخثر. أما الذكر فإن الصبغي Y لديه لا يحمل أي جينات لتكوين عاملي التخثُّر الثامن أو التاسع، لذا فإنه إذا كانت الجينات على الصبغي X بها عيب فإنها ستؤدي إلى ظهور المرض. بما أن الذكر يرث الصبغي X من أمه فإن نسبة إصابة ابن لأم حاملة للمرض هي 50%, أما إذا كانت الأم مصابة فإن نسبة إصابة الابن تصبح 100%. على العكس، الابنة سترث إحدى الصبغيين من الأم والآخر من الأب لذلك فرصة إصابة الذكور بالمرض أكثر من الإناث. حديثًا؛ زادت نسبة إصابة الإناث بالمرض حيث مكّنت طرق العلاج الحديثة والمن متوافرة الذكور من البقاء على قيد الحياة والوصل إلى سن الرشد ليصبحوا آباء وبالتالي زيادة فرص إصابة بناتهم. من الأعراض التي قد تظهر على الإناث غزارة الطمث.
يEوصى بالاختبارات الجينية والاستشارة الوراثية للعائلات المصابة بالناعور. اختبارات ما قبل الولادة، مثل بزل السلى.
وكما هو الحال مع جميع الأمراض الوراثية؛ من الممكن للبشر الإصابة بالمرض من خلال الطفرة، بدلا من وراثته، وذلك بسبب طفرات جديدة في واحدة من أمشاج الأبوين. وتمثل الطفرات التلقائية حوالي 33٪ من جميع حالات الناعور أ، وحوالي 30٪ من حالات الناعور ب. كان من المستحيل تحديد ما إذا كانت الأم هي الحاملة للمرض أم السبب طفرة حتى ظهور اختبار الحمض النووي المباشر الحديث.
شدة المرض
هناك العديد من الطفرات المختلفة التي تسبب كل نوع من الهيموفيليا أ، ب أو ج. بسبب الاختلافات في التغييرات الجينية، مرضى الهيموفيليا لديهم مستوى معيّن من عامل التخثّر. عندما يكون عامل التخثر أقل من 1 % تصنّف الحالة بالهيموفيليا الشديدة. في حال كان من 1-5% تصنّف هذه الحالة بالهيموفيليا المتوسطة أما عندما يكون عامل التخثر طبيعي بين 5-40% تصنّف الحالة بالهيموفيليا المعتدلة.[14]
التشخيص
يمكن للناعور أن يشابه مرض فون ويلبراند.[21] حيث أن انخفاض مستويات عامل فون ويلبراند يمكن أن يؤدي إلى التحلل البروتيني للعامل الثامن. بالإضافة إلى أن الحالات الشديدة من نقص فيتامين K قد تُظهر أعراضًا مشابهة لمرض سيولة الدم. وذلك لأن فيتامين K ضروري لجسم الإنسان لإنتاج عدة عوامل تخثُّر. نقص الفيتامين أمر نادر الحدوث في البالغين والأطفال الأكبر سنًا لكنَّه شائع في حديثي الولادة. يولد الأطفال الرضع لديهم مستويات منخفضة بشكل طبيعي من فيتامين K وليس لديهم حتى الآن بكتيريا الأمعاء التي تقوم بتخليق فيتامين k, يعرف هذا باسم "مرض نزف حديثي الولادة". ولتجنب هذه المضاعفات، يتم حقن حديثي الولادة بصورة روتينية بمكملات فيتامين K.
العلاج
لوقف النزيف يُنصح باستخدام الثلج الموضعي عند حدوث أي نزيف خاصة بالمفاصل مع أخذ مسكنات للآلام. على الرغم من أنه لا يوجد علاج لمرض سيولة الدم، إلا أنه يمكن السيطرة عليه بالحقن العادية لعامل التخثر الناقص، أي العامل الثامن في الناعور أ أو العامل التاسع في الناعور ب. يتم إعطاء الطفل بروتين التجلط المناسب عن طريق الوريد كل 12 ساعة لمدة يومين أو ثلاثة أيام ليساعد الدم علي التجلط وهناك العديد من مشتقات البلازما التي يمكن استخدامها في هذه الحالات وإن كانت بروتينات التجلط التي يتم تصنيعها باستخدام الهندسة الوراثية تعتبر أفضل من البلازما التي قد ينتج عنها انتقال بعض الأمراض مثل التهاب الكبد سي. يكوّن بعض مرضى الهيموفيليا أجسام مضادة (مثبطات) ضد العوامل المُستبدلة المعطاة لهم، وبالتالي فلابد من زيادة كمية العامل لابد من زيادة أو تُعطى المنتجات البديلة غير البشرية، مثل العامل الثامن المُصنَّع من الخنازير. في أوائل عام 2008، وافقت إدارة الغذاء والدواء الأمريكية على (وايث) عامل مضاد للهيموفيليا مُصنَّع بالهندسة الوراثية من جينات من خلايا مبيض الهامستر الصيني. منذ عام 1993 استخدمت على نطاق واسع في الدول الغربية الغنية منتجات عامل التخثر الغير بشرية التي توفر أعلى نقاء وسلامة، لكنَّها مكلفة للغاية فلم تتوافر في العالم النامي. ومن الصعب الحصول على منتجات العوامل من أي نوع في البلدان النامية. [22] وهناك العلاج الوقائي عن طريق حقن الطفل المريض كل 48 ساعة بمعاملات التجلط ورغم أنه يتكلف أكثر من العلاج بالبلازما لكنه يفيد في الحفاظ على المفاصل والعضلات في حالتها الطبيعية حتى لا تحدث أي إعاقة للطفل.[23][24][25]
العلاج الجيني
وأفضل ما يعالج به مريض الهيموفيليا هو العلاج بالجينات مرة واحدة فتكفيه لمدة عام,[26] ويمكن تفادي الانتقال الوراثي للمرض عن طريق إجراء تحاليل قبل الزواج أو الحمل إذا كان الزوج مريضاً أو في حالة وجود طفل مصاب في أسرة الإناث المقبلات على الزواج؛ عليهم قياس نسبة الخلل في الجينات وسرعة التجلط وقياس بروتينات التجلط (8) و(9), في حالة حدوث حمل يتم تحديد نوع الجنين فإذا كان أنثى يستمر الحمل أما إذا كان ذكرًا فيتم عمل تحاليل للدم في الخلايا الموجودة في السائل الأمينوسي وتحليل الجينات حتى نتجنب إعاقة الأطفال.</ref>[27]
تمارين وقائية
من المستحسن أن يقوم الأشخاص المصابين بالهيموفيليا بتمارين محددة لتعزيز المفاصل خاصةً المرفقين، الركبتين والكاحلين. يُوصى بهذه التدريبات يوميًا لتقوية العضلات وبعد النزيف الداخلي خصوصًا لمنع مشاكل النزيف من جديد.[28] من التدريبات: الإحماء، دوائر الكاحل, انحناء الكوع وتمديد عضلات الفخذ.[29]
المحظورات
يُمنع استخدام مضادات التخثر مثل الهيبارين والوارفارين للأشخاص الذين يعانون من الناعور حيث أنها يمكن أن تؤدي إلى تفاقم الحالة. ويُمنع أيضًا الأدوية التي تسبب "سيولة الدم" كآثار جانبية. على سبيل المثال، الأسبرين والايبوبروفين، أو الصوديوم نابروكسين حيث تؤدي إلى إطالة زمن النزيف.[30]
كما يجب تجنب الأنشطة التي يُحتمل التعرض فيها للإصابة، مثل الدراجات النارية والتزلج.[30][31] الرياضات ذات المعدلات العالية من الاتصال الجسدي والإصابات مثل كرة القدم الأميركية والهوكي والملاكمة والمصارعة, كرة القدم والبيسبول وكرة السلة.[30]
علم الأوبئة
الناعور مرض نادر، فقط حوالي 1 في كل 10000 ولادة (أو 1 في كل 5000 من الذكور) الناعور أ 1 في كل 50000 ولادة,[32] الناعور ب 1 من كل 18000 شخص في الولايات المتحدة. كل سنة في الولايات المتحدة، يولد حوالي 400 طفل مصاب بهذا المرض.[33]
تاريخ
اكتشاف علمي
كان أبوالقاسم الزهراوي أول متخصص طبي يصف المرض.[34] في القرن العاشر وصف الأسر التي توفى ذكورها من نزيف بعد صدمات طفيفة. لكن لم يبدأ التحليل العلمي حتى بداية القرن التاسع عشر. عام 1803، كتب طبيب من فيلادلفيا يدعى جون كنارد أوتو مقالة عن "ميل للنزيف يوجد في عائلات معينة".[35] وقد أدرك أن الحالة كانت وراثية وتؤثر على الذكور فقط انتقالًا عبر إناث صحيحات. وكانت ورقته هي الورقة الثانية لوصف خصائص مهمة لاضطراب وراثي مرتبط بالصبغي X (الورقة الأولى كانت في وصف عمى الألوان من قبل جون دالتون الذي درس عائلته). وُصف المرض عام 1813 من قبل جون هاي، حيث نشر مقالة في الصحيفة الطبية بنيو انغلاند.[36][37]
في عام 1924، اكتشف طبيب فنلندي اضطراب نزيف وراثي مماثل لمرض الهيموفيليا في "جزر آلاند"، جنوب غرب فنلندا، [38] ويسمى هذا الاضطراب "مرض فون فيلبراند". وقد ظهرت كلمة "هيموفيليا" أول مرة في وصف هذا المرض سنة 1828م حيث كتبها عالم يدعى هوبف في جامعة زيورخ.[35][39][40] وفي عام 1947 وجد طبيب من بوينس آيرس، أن الناعور نوعين أ وب وهما مرضان منفصلان.
العائلة المالكة الأوروبية

يسمى نزف الدم الوراثي أيضًا بـ"المرض الملكي" لأنه كان سائداً عند عوائل الأُسر الحاكمة. نقلت الملكة فكتوريا [41][42] المرض إلى ابنها، وعن طريق بناتها انتقل هذا المرض إلى العوائل الحاكمة عبر القارات، ومنهم الأُسر الحاكمة في إسبانيا وألمانيا وروسيا.
وزعم راسبوتين أنه كان ناجحًا في علاج الهيموفيليا التي أصيب بها أحد ملوك روسيا، اليكسي ابن نيقولا الثاني. في ذلك الوقت، كانت العلاج مشتركًا من قبل الأطباء وهو استخدام الأسبرين، الذي أدى إلى تفاقم الحالة بدلًا من تقليلها. وببساطة عن طريق تقديم النصح ضد العلاج الطبي، راسبوتين تمكن من تحقيق تحسن ملحوظ وكبير في حالة اليكسي.
في إسبانيا، حفيدة الملكة فيكتوريا من خلال ابنتها الأميرة بياتريس، الأميرة فيكتوريا أوجيني من باتنبرغ أصبحت فيما بعد ملكة إسبانيا. وكان اثنان من أبنائها مصابين بنزف الدم وكلاهما مات من حوادث سيارات بسيطة. توفي ابنها الأكبر، ألفونسو، أمير أستورياس، عن عمر يناهز ال 31 من نزيف داخلي بعد أن اصطدمت سيارته بكشك هاتف. وأيضا أصغر أبنائها، إنفانتي غونزالو، توفي في سن 19 من نزيف في البطن إثر حادث سيارة بسيط حيث اصطدم هو وأخته بجدار أثناء محاولة تجنب دراجة. وتوفى غونزالو بعد يومين من نزيف داخلي دون أن يبدو مصابًا أو يسعى للرعاية الطبية العاجلة.
قضايا تلوث الدم
قبل عام 1985، لم تكن هناك قوانين سنت في الولايات المتحدة لفحص الدم. ونتيجة لذلك، كان الكثير من الناس المصابين بالناعور في خطر شديد للإصابة بفيروس نقص المناعة البشرية والتهاب الكبد الوبائي عن طريق منتجات الدم. وتشير التقديرات إلى أن أكثر من 50٪ من السكان المصابين بالناعور، أي أكثر من 10,000 شخص، أصيبوا بفيروس نقص المناعة البشرية في الولايات المتحدة وحدها. [43] في أواخر السبعينات وأوائل / منتصف الثمانينات، وُضعت أساليب جديدة في إنتاج عوامل التجلط. مثل البسترة والمعالجة الحرارية.
انظر أيضًا
المصادر
- علم الأحياء بواسطة بيتر هـ. ريفن،جورج ب. جونسون،جوناثان ب. لوسوس،كينيث أ. ماسون،سوزان ر. سنجر، صفحة 240. نسخة محفوظة 2 يناير 2020 على موقع واي باك مشين.
- المعجم الطبي. نسخة محفوظة 12 يونيو 2018 على موقع واي باك مشين.
- "What Is Hemophilia? - NHLBI, NIH". www.nhlbi.nih.gov. مؤرشف من الأصل في 02 يوليو 2016. اطلع عليه بتاريخ 21 يونيو 2016. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Hemophilia A: MedlinePlus Medical Encyclopedia". www.nlm.nih.gov. مؤرشف من الأصل في 05 يوليو 2016. اطلع عليه بتاريخ 21 يونيو 2016. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Prasad Mathew, MBBS, DCH, eMedicine - Hemophilia C نسخة محفوظة 2008-12-02 على موقع واي باك مشين.
- Páramo, Laura; Enciso Olivera, Leonardo Jose; Noreña, Ivan; Amaya, María A.; Santacruz, Juan C. (2019-03-05). "First Case of Acquired Hemophilia B in a Patient with HIV Infection: Case Report and Literature Review". Cureus. 11 (3): e4179. doi:10.7759/cureus.4179. ISSN 2168-8184. PMC 6504016. PMID 31106079. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Hemophilia B: MedlinePlus Medical Encyclopedia". www.nlm.nih.gov. مؤرشف من الأصل في 05 يوليو 2016. اطلع عليه بتاريخ 21 يونيو 2016. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Franchini, M; Mannucci, PM (December 2013). "Acquired haemophilia A: a 2013 update". Thrombosis and Haemostasis. 110 (6): 1114–20. CiteSeerX = 10.1.1.684.7962 10.1.1.684.7962. doi:10.1160/TH13-05-0363. PMID 24008306. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Pool, Judith G.; Hershgold, Edwabd J.; Pappenhagen, Albert R. (July 1964). "High-potency Antihæmophilic Factor Concentrate prepared from Cryoglobulin Precipitate". Nature. 203 (4942): 312. Bibcode:1964Natur.203..312P. doi:10.1038/203312a0. ISSN 0028-0836. PMID 14201780. مؤرشف من الأصل في 14 مارس 2020. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "How Is Hemophilia Treated?". NHLBI. July 13, 2013. مؤرشف من الأصل في 17 سبتمبر 2016. اطلع عليه بتاريخ 10 سبتمبر 2016. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Manco-Johnson MJ, Abshire TC, Shapiro AD, Riske B, Hacker MR, Kilcoyne R, Ingram JD, Manco-Johnson ML, Funk S, Jacobson L, Valentino LA, Hoots WK, Buchanan GR, DiMichele D, Recht M, Brown D, Leissinger C, Bleak S, Cohen A, Mathew P, Matsunaga A, Medeiros D, Nugent D, Thomas GA, Thompson AA, McRedmond K, Soucie JM, Austin H, Evatt BL (2007). "Prophylaxis versus episodic treatment to prevent joint disease in boys with severe hemophilia". N. Engl. J. Med. 357 (6): 535–544. doi:10.1056/NEJMoa067659. PMID 17687129. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Roosendaal G, Lafeber F (2007). "Prophylactic treatment for prevention of joint disease in hemophilia—cost versus benefit". N. Engl. J. Med. 357 (6): 603–605. doi:10.1056/NEJMe078098. PMID 17687136. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Data Collection - WFH Annual Global Survey - World Federation of Hemophilia". www.wfh.org. مؤرشف من الأصل في 25 يوليو 2019. اطلع عليه بتاريخ 10 ديسمبر 2018. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Hemophilia Overview eMedicine from webMD. Dimitrios P Agaliotis, MD, PhD, FACP, Robert A Zaiden, MD, Fellow, and Saduman Ozturk, PA-C. Updated: 24 November 2009. "نسخة مؤرشفة". Archived from the original on 12 أبريل 2011. اطلع عليه بتاريخ 31 مايو 2016. الوسيط
|CitationClass=تم تجاهله (مساعدة)صيانة CS1: BOT: original-url status unknown (link) - Hemophilia Complications Mayo Clinic Staff. 16 May 2009 نسخة محفوظة 9 نوفمبر 2013 على موقع واي باك مشين.
- Rodriguez-Merchan, E. Carlos. "Musculoskeletal Complications of Hemophilia". مؤرشف من الأصل في 17 ديسمبر 2019. اطلع عليه بتاريخ 04 أبريل 2014. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Valentino LA, Hakobyan N, Rodriguez N, Hoots WK; Kakobyan, N; Rodriguez, N; Hoots, WK (November 2007). "Pathogenesis of haemophilic synovitis: experimental studies on blood-induced joint damage". Haemophilia. 13 Suppl 3: 10–3. doi:10.1111/j.1365-2516.2007.01534.x. PMID 17822515. مؤرشف من الأصل في 23 نوفمبر 2018. اطلع عليه بتاريخ 04 أبريل 2014. الوسيط
|CitationClass=تم تجاهله (مساعدة)صيانة CS1: أسماء متعددة: قائمة المؤلفون (link) - World Federation of Hemophilia Frequently Asked Questions. 2005 نسخة محفوظة 27 يوليو 2016 على موقع واي باك مشين. [وصلة مكسورة]
- "2008 Global Survey Report" (PDF). الوسيط
|CitationClass=تم تجاهله (مساعدة);|archive-url=is malformed: timestamp (مساعدة) - Prasad Mathew, MBBS, DCH, eMedicine - Hemophilia C نسخة محفوظة 02 ديسمبر 2008 على موقع واي باك مشين.
- "Molecular basis of von Willebrand disease and its clinical implications". Haematologica. 89 (9): 1036. 1 September 2004. PMID 15377463. مؤرشف من الأصل (PDF) في 02 أكتوبر 2011. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Manco-Johnson MJ, Abshire TC, Shapiro AD, Riske B, Hacker MR, Kilcoyne R, Ingram JD, Manco-Johnson ML, Funk S, Jacobson L, Valentino LA, Hoots WK, Buchanan GR, DiMichele D, Recht M, Brown D, Leissinger C, Bleak S, Cohen A, Mathew P, Matsunaga A, Medeiros D, Nugent D, Thomas GA, Thompson AA, McRedmond K, Soucie JM, Austin H, Evatt BL (2007). "Prophylaxis versus episodic treatment to prevent joint disease in boys with severe hemophilia". N. Engl. J. Med. 357 (6): 535–544. doi:10.1056/NEJMoa067659. PMID 17687129. مؤرشف من الأصل في 16 ديسمبر 2019. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Roosendaal G, Lafeber F (2007). "Prophylactic treatment for prevention of joint disease in hemophilia—cost versus benefit". N. Engl. J. Med. 357 (6): 603–605. doi:10.1056/NEJMe078098. PMID 17687136. مؤرشف من الأصل في 18 مارس 2009. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Kogenate Product Approval InformationUSDA Center for Biologics Evaluation and Research. نسخة محفوظة 12 مايو 2009 على موقع واي باك مشين. "نسخة مؤرشفة". Archived from the original on 12 مايو 2009. اطلع عليه بتاريخ 20 يوليو 2018. الوسيط
|CitationClass=تم تجاهله (مساعدة)صيانة CS1: BOT: original-url status unknown (link) - Hayes, P. 2009. FDA Approves Kogenate For Prophylaxis. Hemaware: the bleeding disorder's magazine. National Hemophilia Foundation. March/April 2009. Vol 14, Issue 2. p. 18. نسخة محفوظة 09 يوليو 2018 على موقع واي باك مشين.
- Nicholas Wade (10 December 2011). "Treatment for Blood Disease Is Gene Therapy Landmark". نيويورك تايمز. مؤرشف من الأصل في 12 أكتوبر 2017. الوسيط
|CitationClass=تم تجاهله (مساعدة) - (20 November 2014) Gene therapy provides safe, long-term relief for patients with severe hemophilia B Science Daily, Retrieved 17 December 2014 نسخة محفوظة 03 يوليو 2017 على موقع واي باك مشين.
- Mulder, K. 2006. Exercises for People with Hemophilia. World Federation of Hemophilia [وصلة مكسورة] نسخة محفوظة 15 أبريل 2012 على موقع واي باك مشين.
- (PDF) https://web.archive.org/web/20120415011734/http://www.wfh.org/2/docs/Publications/General_Guides/Exercise_Guide_med.pdf. مؤرشف من الأصل (PDF) في 15 أبريل 2012. الوسيط
|CitationClass=تم تجاهله (مساعدة); مفقود أو فارغ|title=(مساعدة) - How to Deal with Hemophilia Reviewed by: Larissa Hirsch, MD 2007. kidshealth.org by Nemours. Retrieved 23 January 2010. نسخة محفوظة 02 ديسمبر 2016 على موقع واي باك مشين.
- "Playing it Safe: Bleeding Disorders, Sports and Exercise". Booklet. National Hemophilia Foundation. نسخة محفوظة 15 ديسمبر 2012 على موقع واي باك مشين.
- World Federation of Hemophilia Frequently Asked Questions About Hemophilia نسخة محفوظة 12 يونيو 2008 على موقع واي باك مشين. "نسخة مؤرشفة". Archived from the original on 6 مارس 2016. اطلع عليه بتاريخ 28 سبتمبر 2020. الوسيط
|CitationClass=تم تجاهله (مساعدة)صيانة CS1: BOT: original-url status unknown (link) - "U.S. National Library of Medicine". مؤرشف من الأصل في 5 يوليو 2016. اطلع عليه بتاريخ 02 ديسمبر 2007. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Case of the Week 175". University of Utah Medical Library. مؤرشف من الأصل في 19 مايو 2011. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Nilsson IM (1994). "Haemophilia--then and now". Sydsvenska medicinhistoriska sallskapets arsskrift. 31: 33–52. PMID 11640407. الوسيط
|CitationClass=تم تجاهله (مساعدة) - DIGITISED EARLY PAPERS AND BOOKS ON HUMAN AND MEDICAL GENETICS Genetics and Medicine Historical Network, Cardiff University. نسخة محفوظة 25 أبريل 2012 على موقع واي باك مشين.
- Hay J (July 1813). "Account of a remarkable hæmorrhagic disposition, existing in many individuals of the same family". N Engl J Med Surg. 2 (3): 221–5. doi:10.1056/NEJM181307010020302. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Haemophilia Special Issue: von Willebrand's Disease: a Report from a Meeting in the Åland Islands". مؤرشف من الأصل في 29 يونيو 2017. اطلع عليه بتاريخ 22 نوفمبر 2012. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "The History of hemophilia". مؤرشف من الأصل في 15 أبريل 2012. اطلع عليه بتاريخ 05 يونيو 2009. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Chapter 38 Coagulation Factors V and VIII by GC White and GE Gilbert in "Blood: principles and practice of hematology: 2nd edition" 2003. Eds. Robert I. Handin, Samuel E. Lux, Thomas P. Stossel. ISBN 978-0-7817-1993-3 نسخة محفوظة 03 مايو 2016 على موقع واي باك مشين.
- Michael Price (8 October 2009). "Case Closed: Famous Royals Suffered From Hemophilia". ScienceNOW Daily News. AAAS. مؤرشف من الأصل في 12 أكتوبر 2009. اطلع عليه بتاريخ 09 أكتوبر 2009. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Evgeny I. Rogaev; et al. (8 October 2009). "Genotype Analysis Identifies the Cause of the "Royal Disease"". Science. مؤرشف من الأصل في 14 أكتوبر 2009. اطلع عليه بتاريخ 09 أكتوبر 2009. الوسيط
|CitationClass=تم تجاهله (مساعدة) - In re Rhone-Poulenc Rorer Inc., 51 F.3d 1293, 1296 (7th Cir. 1995), Projectposner.org, Retrieved 28 January 2008 نسخة محفوظة 04 مارس 2016 على موقع واي باك مشين.
 بوابة طب
بوابة طب
