هيموفيليا أ
هيموفيليا أ (بالإنجليزية: Haemophilia A) هو نقص وراثي في عامل التخثر الثامن، مما يسبب زيادة النزف، وعادة ما يصيب الذكور. ويتم توارثه في معظم الحالات باعتباره سمة متنحية مرتبطة بالكروموسوم X، على الرغم من أن هناك حالات تنشأ عن الطفرات العفوية.[1][2]
| هيموفيليا أ | |
|---|---|
| معلومات عامة | |
| الاختصاص | علم الدم |
| من أنواع | هيموفيليا ، ومرض متنح مرتبط بالجنس ، واعتلال خثري |
| الأسباب | |
| الأسباب | نقص العامل الثامن[1] |
| المظهر السريري | |
| المضاعفات | يجب وضع لقاح التهاب الكبد الفيروسي ب في الاعتبار[1] |
| الإدارة | |
| التشخيص | زمن النزف[1] |
| العلاج | تركيزات العامل الثامن[1] |
ويمكن استخدام العامل الثامن كدواء لعلاج ومنع النزيف لدى الأشخاص المصابين بالهيموفيليا أ.[3]
العلامات والأعراض
من حيث أعراض الهيموفيليا أ، فهناك نوبات نزيف داخلي أو خارجي. ويعاني الأفراد المصابون بالهيموفيليا الشديدة من نزيف أكثر حدة وتكرارا، في حين يعاني الآخرون المصابون بالهيموفيليا البسيطة من أعراض طفيفة باستثناء بعد الجراحة أو الرضة الخطيرة. بينما الهيموفيليا المعتدلة لها أعراض متفاوتة بين الأشكال الشديدة والبسيطة.[4]
النزيف المطول من الوخز بالإبر هو علامة أخرى شائعة ومبكرة للهيموفيليا، وقد تؤدي هذه العلامات إلى القيام باختبارات الدم التي تشير إلى الهيموفيليا.[5] في أشخاص آخرين، وخاصة أولئك الذين يعانون من الهيموفيليا البسيطة أو المعتدلة، قد تؤدي أي رضة إلى أول نزيف خطير. وتسبب الهيموفيليا زيادة شديدة في خطر النزف لفترات طويلة بعد الإصابات الشائعة، وقد يكون النزيف عفوي وبدون سبب واضح في الحالات الشديدة. وقد يحدث النزف في أي مكان في الجسم، ويطول زمن النزف السطحي مثل ذلك الذي تسببه السحجات أو التمزقات السطحية، ويمكن تكسير قشرة التئام الجرح بسهولة بسبب نقص الفيبرين، مما قد يسبب إعادة النزف.[4] ومن الأماكن الأكثر خطورة للنزف:[6]
يدل النزف العضلي ونزف المفاصل (تدمي المفصل) على الهيموفيليا،[7] في حين أن نزف الجهاز الهضمي والنزيف الدماغي يكون ذات صلة باضطرابات التخثر الأخرى. وعلى الرغم من أن النزف المفصلي ليس مهددا للحياة، إلا أنه من أخطر أعراض الهيموفيليا. فقد يسبب النزيف المتكرر في محفظة المفصل تلف دائم وتشوه للمفصل، مما يؤدي إلى التهاب المفاصل المزمن والعجز. لا يحدث تلف المفصل نتيجة وجود دم في المحفظة، ولكن بسبب عملية الالتئام. عندما يتم تكسير الدم الموجود في المفصل بواسطة إنزيمات الجسم، يتم أيضا تكسير العظام في تلك المنطقة، مما يسبب الكثير من الألم للشخص المصاب بالمرض.
المضاعفات
أحد المعضلات العلاجية هو تطوير الأجسام المضادة المثبطة ضد العامل الثامن بسبب الضخ المتكرر. وتتطور هذه الأجسام المضادة بسبب أن الجسم يتعرف على "العامل الثامن الطبيعي" كجسم غريب؛ لأن الجسم ليس لديه نسخة خاصة به. وتكمن المشكلة في هؤلاء الأفراد أن ضخ العامل الثامن يكون غير فعال. ويصبح العامل السابع الذي يتم تنشيطه حديثا متاحا لعلاج النزف لدى الأفراد الذين يعانون من الهيموفيليا ومثبطات العوامل.[4][8]
علم الوراثة
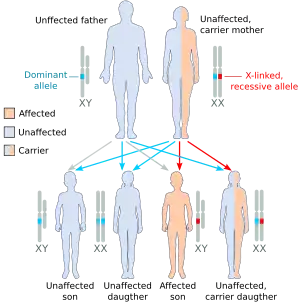
تورث هيموفيليا أ كصفة متنحية مرتبطة بالكروموسوم X، وتحدث في الذكور وفي الإناث متماثلة الزيجوت (تحدث فقط في ذرية الإناث الحاملة للمرض والذكور المصابة[9]). ومن المعروف أن هيموفيليا أ البسيطة تحدث في الإناث فردانية الزيجوت بسبب تعطيل الكروموسوم X، لذلك فمن المستحسن قياس مستويات العامل الثامن والتاسع في جميع حاملات المرض المعروفة أو المحتملة قبل الجراحة وفي حالة النزيف الشديد إكلينيكيا.[4][10]
يصاب حوالي 5-10٪ من الأشخاص بهيموفيليا أ؛ بسبب امتلاكهم نسخة مختلة من بروتين العامل الثامن، في حين أن الباقي يصابون؛ لأنهم ينتجون العامل الثامن بكميات غير كافية (نقص كمي).[10] ومن بين أولئك الذين يعانون من نقص حاد بسبب نشاط 1٪ فقط من العامل الثامن، يمتلك 45-50٪ منهم نفس الطفرة، وهي الانعكاس داخل جين العامل الثامن، الذي يؤدي إلى الحد على إنتاج البروتين.[10]
وبما أن كلا من أشكال الهيموفيليا يمكن أن تكون ناجمة عن مجموعة متنوعة من الطفرات المختلفة، فإن التشخيص الأولي والتصنيف يتم عن طريق قياس نشاط البروتين بدلا من الاختبارات الجينية، على الرغم من أن الاختبارات الجينية يوصى بها لاختبار أفراد الأسرة في حالة معرفة إصابة أحدهم بالهيموفيليا.[4][10] حوالي 30٪ من المرضى ليس لديهم تاريخ عائلي للمرض، ومن المفترض أن يكون مرضهم نتيجة طفرات جديدة.[11]
التشخيص
قد يشتبه في تشخيص هيموفيليا أ عندما يكشف اختبار التخثر عن زيادة زمن الثرمبوبلاستين الجزئي مع زمن البروثرومبين الطبيعي وزمن النزف الطبيعي. اختبار زمن الثرومبوبلاستين الجزئي هو أول اختبار دم يتم عمله عند الاشتباه في الهيموفيليا.[12] ويتم التشخيص بوجود مستويات منخفضة جدا من العامل الثامن. وعادة ما يكون التاريخ العائلي للمرض موجودا، وإن لم يكن ضروريا. وفي الآونة الأخيرة، يتم إجراء الاختبارات الجينية المتاحة لتحديد نسبة خطر إصابة الفرد بالهيموفيليا. ويشمل تشخيص هيموفيليا أ أيضا مستوى الشدة، التي يمكن أن تتراوح من بسيطة إلى شديدة استنادا إلى كمية العامل النشط والعامل الموجود في الدم. ولا تتغير مستويات العامل الثامن عادة خلال حياة الفرد. هيموفيليا أ الشديدة هي الشكل الأكثر شيوعا الذي يحدث في غالبية الأشخاص المتضررين. وغالبا ما يعاني الأفراد المصابون بالهيموفيليا البسيطة من نوبات نزيف قليلة أو من عدم وجود نوبات نزيف إلا في حالة الرضة الخطيرة، مثلما يحدث مع خلع الأسنان أو الجراحة.[4]
الخطورة
هناك العديد من الطفرات المختلفة التي تسبب هيموفيليا أ. وبسبب الاختلافات في التغيرات في الجين المعني (والبروتين الناتج)، فغالبا ما يكون لدى الأفراد الذين يعانون من الهيموفيليا مستوى ما من عامل التخثر النشط. ويصنف الأفراد الذين لديهم نسبة العامل النشط أقل من 1٪ على أنهم يعانون من الهيموفيليا الشديدة، والذين لديهم عامل نشط بنسبة 1-5٪ لديهم هيموفيليا معتدلة، والذين يعانون من هيموفيليا بسيطة لديهم ما بين 5-40٪ من المستويات الطبيعية لعامل التخثر النشط.[13]
التشخيص التفريقي
هناك اثنان من التشخيصات التفريقية الأكثر شيوعا وهما هيموفيليا ب (وهو نقص في العامل التاسع)، ومرض فون ويلبراند (وهو نقص في عامل فون ويل براند اللازم لسلامة عمل العامل الثامن[14])، كما تعتبر الهيموفيليا ج أيضا واحدة من التشخيصات التفريقية.[2]
العلاج
فيما يتعلق بمعالجة هذا الاضطراب الوراثي، فإن معظم الأفراد الذين يعانون من الهيموفيليا الشديدة يحتاجون إلى مكملات منتظمة مع العامل الثامن معاد التركيب أو مركز من البلازما عن طريق الوريد. ونظام العلاج الوقائي متغير للغاية ومحدد بشكل فردي.[6] وفي الأطفال، يمكن الوصول إلى مدخل سهل للوريد[15] لتقليل القنية المتكررة فيه. وقد جعلت هذه الوسائل الوقاية في الهيموفيليا أسهل بكثير للأسر؛ للحد من مشاكل العثور على الوريد للضخ عدة مرات في الأسبوع. ومع ذلك، هناك مخاطر تنطوي على استخدامها، والأكثر إثارة للقلق هو العدوى، فالدراسات تختلف ولكن بعضها يبين أن معدلات العدوى عالية.[16] وعادة يمكن علاج هذه العدوى بالمضادات الحيوية في الوريد، ولكن في بعض الأحيان يجب إزالة الجهاز،[17] وهناك أيضا دراسات أخرى تظهر خطر تكون الجلطات في طرف القسطرة. ويعالج بعض الأفراد الذين يعانون من هيموفيليا شديدة ومعظم المصابين بهيموفيليا معتدلة وبسيطة فقط حسب الحاجة بدون جدول وقائي منتظم.[18] وغالبا ما يُعالَج المصابون بهيموفيليا بسيطة في كثير من الأحيان بالدزموبريسين، الذي يطلق العامل الثامن المخزَن من جدران الأوعية الدموية.[19]
توقع سير المرض
قد اتبعت دراستان هولنديتان مرضى الهيموفيليا لعدة سنوات.[23][24] ووجدت كلتا الدراستين أن العدوى الفيروسية شائعة في الهيموفيليا؛ بسبب عمليات نقل الدم المتكررة التي تعرضهم لخطر الإصابة بالعدوى المنقولة بالدم، مثل فيروس نقص المناعة البشرية والتهاب الكبد C. وفي آخر دراسة تابعت المرضى من عام 1992 إلى عام 2001، كان متوسط العمر المتوقع للذكور 59 عاما. وإذا استُبعدت الحالات المعروفة بالإصابة الفيروسية، كان متوسط العمر المتوقع 72 عاما، وهو قريب من متوسط عمر عامة السكان. توفي 26٪ من الحالات من الإيدز، و22٪ من التهاب الكبد C.[24]
وبائيات
تحدث الهيموفيليا أ في حوالي 1 لكل 5000 ذكر،[10] في حين أن نسبة الإصابة بالهيموفيليا ب هي 1 لكل 30,000 ذكر، 85٪ منهم لديهم الهيموفيليا أ و15٪ لديهم الهيموفيليا ب.[10]
المراجع
- "Hemophilia A: MedlinePlus Medical Encyclopedia". www.nlm.nih.gov. مؤرشف من الأصل في 05 يوليو 2016. اطلع عليه بتاريخ 24 يونيو 2016. الوسيط
|CitationClass=تم تجاهله (مساعدة); تحقق من التاريخ في:|تاريخ أرشيف=(مساعدة) - "Haemophilia A (Factor VIII Deficiency) information | Patient". Patient. مؤرشف من الأصل في 27 أكتوبر 2018. اطلع عليه بتاريخ 24 يونيو 2016. الوسيط
|CitationClass=تم تجاهله (مساعدة) - WHO Model Formulary 2008 (PDF). World Health Organization. 2009. صفحات 259–260. ISBN 9789241547659. مؤرشف من الأصل (PDF) في 9 ديسمبر 2019. اطلع عليه بتاريخ 08 يناير 2017. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Konkle, Barbara A.; Josephson, Neil C.; Nakaya Fletcher, Shelley (1993-01-01). Pagon, Roberta A.; Adam, Margaret P.; Ardinger, Holly H.; Wallace, Stephanie E.; Amemiya, Anne; Bean, Lora JH; Bird, Thomas D.; Fong, Chin-To; Mefford, Heather C. (المحررون). Hemophilia A. Seattle (WA): University of Washington, Seattle. PMID 20301578. مؤرشف من الأصل في 19 أكتوبر 2019. الوسيط
|CitationClass=تم تجاهله (مساعدة)update 2014 - Lissauer, Tom; Fanaroff, Avroy A.; Miall, Lawrence; Fanaroff, Jonathan (2015-06-10). Neonatology at a Glance (باللغة الإنجليزية). John Wiley & Sons. صفحة 135. ISBN 9781118767429. مؤرشف من الأصل في 14 يوليو 2018. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "How Is Hemophilia Treated? - NHLBI, NIH". www.nhlbi.nih.gov. مؤرشف من الأصل في 05 أكتوبر 2017. اطلع عليه بتاريخ 08 يوليو 2016. الوسيط
|CitationClass=تم تجاهله (مساعدة); تحقق من التاريخ في:|تاريخ أرشيف=(مساعدة) - "Management of Joint Bleeding in Hemophilia: Management of Acute Hemarthrosis". Medscape. Medscape.org. مؤرشف من الأصل في 06 يوليو 2017. اطلع عليه بتاريخ 01 أغسطس 2016. الوسيط
|CitationClass=تم تجاهله (مساعدة); تحقق من التاريخ في:|تاريخ أرشيف=(مساعدة) - Ma, Alice D.; Carrizosa, Daniel (2006-01-01). "Acquired Factor VIII Inhibitors: Pathophysiology and Treatment". ASH Education Program Book (باللغة الإنجليزية). 2006 (1): 432–437. doi:10.1182/asheducation-2006.1.432. ISSN 1520-4391. PMID 17124095. مؤرشف من الأصل في 2 يونيو 2018. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Nair, Preethi S.; Shetty, S.; Ghosh, Kanjaksha (2012-01-01). "A homozygous female hemophilia A". Indian Journal of Human Genetics. 18 (1): 134–136. doi:10.4103/0971-6866.96685. ISSN 0971-6866. PMC 3385172. PMID 22754241. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Kliegman, Robert (2011). Nelson textbook of pediatrics (الطبعة 19th). Philadelphia: Saunders. صفحات 1700–1. ISBN 978-1-4377-0755-7. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Bowen, D J (2002). "Haemophilia A and haemophilia B: molecular insights". Molecular Pathology. 55 (1): 1–18. doi:10.1136/mp.55.1.1. PMC 1187139. PMID 11836440. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "OMIM Entry - # 306700 - HEMOPHILIA A; HEMA". omim.org. مؤرشف من الأصل في 15 مايو 2019. اطلع عليه بتاريخ 08 يوليو 2016. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Hemophilia A في موقع إي ميديسين
- "Von Willebrand's Disease. About Von Willebrand's Disease | Patient". Patient (باللغة الإنجليزية). مؤرشف من الأصل في 27 أكتوبر 2018. اطلع عليه بتاريخ 08 يوليو 2016. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Ma, Alice D.; Roberts, Harold R.; Escobar, Miguel A. (2012-10-03). Hemophilia and Hemostasis: A Case-Based Approach to Management (باللغة الإنجليزية). John Wiley & Sons. ISBN 9781118439302. مؤرشف من الأصل في 06 يونيو 2020. الوسيط
|CitationClass=تم تجاهله (مساعدة)Google books no page - Santagostino, Elena; Mancuso, Maria Elisa (2008-09-01). "Barriers to primary prophylaxis in haemophilic children: the issue of the venous access". Blood Transfusion. 6 (Suppl 2): s12–s16. doi:10.2450/2008.0031-08. ISSN 1723-2007. PMC 2652218. PMID 19105504. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Guidelines for the Prevention of Intravascular Catheter-Related Infections". www.cdc.gov. مؤرشف من الأصل في 02 أكتوبر 2018. الوسيط
|CitationClass=تم تجاهله (مساعدة); تحقق من التاريخ في:|تاريخ أرشيف=(مساعدة) - Ljung, Rolf (2007-09-01). "The risk associated with indwelling catheters in children with haemophilia". British Journal of Haematology (باللغة الإنجليزية). 138 (5): 580–586. doi:10.1111/j.1365-2141.2007.06703.x. ISSN 1365-2141. PMID 17686052. مؤرشف من الأصل في 24 سبتمبر 2017. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Franchini, Massimo; Lippi, Giuseppe (2011-10-01). "The use of desmopressin in acquired haemophilia A: a systematic review". Blood Transfusion. 9 (4): 377–382. doi:10.2450/2011.0113-10. ISSN 1723-2007. PMC 3200405. PMID 21839010. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Groundbreaking gene therapy trial set to cure haemophilia A". Barts Health NHS Trust. 14 December 2017. مؤرشف من الأصل في 15 ديسمبر 2017. اطلع عليه بتاريخ 14 ديسمبر 2017. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Haemophilia A trial results 'mind-blowing'". BBC. 14 December 2017. مؤرشف من الأصل في 08 مارس 2019. اطلع عليه بتاريخ 14 ديسمبر 2017. الوسيط
|CitationClass=تم تجاهله (مساعدة); تحقق من التاريخ في:|تاريخ أرشيف=(مساعدة) - "AAV5–Factor VIII Gene Transfer in Severe Hemophilia A". New England Journal of Medicine. 16 December 2017. مؤرشف من الأصل في 16 مايو 2019. اطلع عليه بتاريخ 14 ديسمبر 2017. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Triemstra, Mattanja (1995-12-01). "Mortality in Patients with Hemophilia: Changes in a Dutch Population from 1986 to 1992 and 1973 to 1986". Annals of Internal Medicine. 123 (11): 823. doi:10.7326/0003-4819-123-11-199512010-00002. ISSN 0003-4819. مؤرشف من الأصل في 8 أبريل 2020. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Plug, I.; Van Der Bom, J. G.; Peters, M.; Mauser-Bunschoten, E. P.; De Goede-Bolder, A.; Heijnen, L.; Smit, C.; Willemse, J.; Rosendaal, F. R. (2006-03-01). "Mortality and causes of death in patients with hemophilia, 1992–2001: a prospective cohort study1". Journal of Thrombosis and Haemostasis (باللغة الإنجليزية). 4 (3): 510–516. doi:10.1111/j.1538-7836.2006.01808.x. ISSN 1538-7836. مؤرشف من الأصل في 13 فبراير 2018. الوسيط
|CitationClass=تم تجاهله (مساعدة)
لمزيد من القراءة
- Casana, P.; Cabrera, N.; Cid, A. R.; Haya, S.; Beneyto, M.; Espinos, C.; Cortina, V.; Dasi, M. A.; Aznar, J. A. (2008-07-01). "Severe and moderate hemophilia A: identification of 38 new genetic alterations". Haematologica (باللغة الإنجليزية). 93 (7): 1091–1094. doi:10.3324/haematol.12344. ISSN 0390-6078. PMID 18403393. مؤرشف من الأصل في 01 أغسطس 2018. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Roberts, Harold R. Haemophilia and Haemostasis: A Case-based Approach to Management (باللغة الإنجليزية). John Wiley & Sons. ISBN 9780470766439. مؤرشف من الأصل في 13 أغسطس 2020. اطلع عليه بتاريخ 08 يوليو 2016. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Collins, Peter; Baudo, Francesco; Huth-Kühne, Angela; Ingerslev, Jørgen; Kessler, Craig M; Castellano, Maria E Mingot; Shima, Midori; St-Louis, Jean; Lévesque, Hervé (7 June 2010). "Consensus recommendations for the diagnosis and treatment of acquired hemophilia A". BMC Research Notes. 3: 161. doi:10.1186/1756-0500-3-161. ISSN 1756-0500. PMC 2896368. PMID 20529258. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Coppola, Antonio; Windyga, Jerzy; Tufano, Antonella; Yeung, Cindy; Di Minno, Matteo Nicola Dario (9 February 2015). "Treatment for preventing bleeding in people with haemophilia or other congenital bleeding disorders undergoing surgery". Cochrane Database of Systematic Reviews (باللغة الإنجليزية). John Wiley & Sons, Ltd. doi:10.1002/14651858.cd009961.pub2. مؤرشف من الأصل في 10 فبراير 2018. اطلع عليه بتاريخ 12 يوليو 2016. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Kellogg, TF (November 1974). "Steroid balance and tissue cholesterol accumulation in germfree and conventional rats fed diets containing saturated and polyunsaturated fats". Journal of Lipid Research. 15 (6): 574–9. ISSN 0253-0716. PMID 4430880. مؤرشف من الأصل في 13 أغسطس 2020. اطلع عليه بتاريخ 26 ديسمبر 2017. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Armstrong, Elina; Hillarp, Andreas. "Assay discrepancy in mild haemophilia A". European Journal of Haematology. Supplementum. 76: 48–50. doi:10.1111/ejh.12374. ISSN 0902-4506. مؤرشف من الأصل في 22 مايو 2018. اطلع عليه بتاريخ 26 ديسمبر 2017. الوسيط
|CitationClass=تم تجاهله (مساعدة)
 بوابة طب
بوابة طب
