تكون العظم الناقص
تكوّن العظم الناقص (بالإنجليزية osteogenesis imperfecta) ويختصر (OI) وله أسماء عديدة أخرى مثل: "متلازمة لوبشتاين"،[1] و"تخلق العظم الناقص"، و"العظم الزجاجي"؛ هو مرض وراثي في أغلب حالاته،.[2] أي يكفي أن يكون أحد الوالدين حاملا للمرض فيصاب أحد الأبناء به. ولكن في حالات قليلة قد يكون سبب الإصابة هو طفرة جينية نتيجة الخلل في الكولاجين من النوع الأول،[3] الذي يعد مصدر البروتين الأساسي في بنية العظم. ولكن يعتبر العامل الوراثي هو الأكثر شيوعاً ويشكل حوالي 80 إلى 85% من أسباب الإصابة بمرض تكون العظم الناقص.[4] إن اضطراب العظام الخلاقي يترافق مع عيب في النسيج الضام، مما يؤدي إلى عدم القدرة على بناء العظم أو تكوينه ويؤدي إلى سهولة كسر العظام، وغالباً ما يكون سبب تلك الكسور غير ظاهر. والأشخاص المصابون بهذا المرض غالبا ما يكون لديهم ضعف في العضلات وليونة في المفاصل وتشوهات في الهيكل العظمي، مما يؤدي إلى إعاقة في الحركة وتأخر في ممارسه الحياة الطبيعية. وفي مرحلة المراهقة تكون العظام بشكل عام أقل عرضة للكسر.[5]
| تكوّن العظم الناقص | |
|---|---|
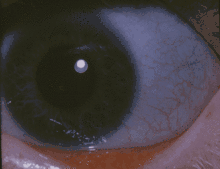
 تشكل الزرقاء الصلبة في العين لشخص مصاب بتكون العظم الناقص. تشكل الزرقاء الصلبة في العين لشخص مصاب بتكون العظم الناقص. | |
| معلومات عامة | |
| الاختصاص | علم الوراثة الطبية |
| من أنواع | خلل تنسج عظمي غضروفي |
الأشخاص المصابون بالنوع الأول (I) تكون نصف كمية الكولاجين وبنيتها في أجسادهم طبيعية. أما الأشخاص المصابون بالأنواع الأخرى (II,III,IV) فيكون الكولاجين في أجسادهم غير سليم من حيث الكميه والبنية.[6]
الأسباب
في أغلب حالات الإصابة بالمرض، يكون العامل الوراثي هو السبب.[7] أما باقي الحالات فقد يكون سببها خلل جيني تكون فيه العظام هشة وسهلة الكسر.[8] لا يوجد علاج لهذا المرض، ولكن تصبح العظام بشكل عام أقل عرضة للكسر في سن المراهقة (سن البلوغ).[9] هذا النقص ينشأ من إحلال الجلايسين لأضخم الأحماض الأمينية في الكولاجين الثلاثي الحلزوني الهيكل، مما يشكل سلاسل في الجانب الأكبر من الأحماض الأمينية وقد يخلق عائقا ويشكل انتفاخا في مجمع الكولاجين، والذي بدوره يؤثر على كل من الحركيات الجزيئية (بالإنجليزية: nanomechanics) فضلا عن التفاعل بين الجزيئات الأخرى، وكلتا الحالتين تشكلان خطرا. ونتيجة لذلك، قد يكون ردة فعل الجسم تحليل بنية الكولاجين غير الطبيعية. وإذا كان الجسم لا يستطيع تدمير الكولاجين بشكل طبيعي، يتم تبديل العلاقة بين ألياف الكولاجين وبلورات هيدروكسيباتيت، المشكّلة للعظام، مما يسبب هشاشة فيه.[6][10] وبهذا يمكن القول أن أسباب ظاهرة تكوّن العظم الناقص عديدة تبدأ من الوراثة حتى طفرات الجينات والصبغيات ومستوى عمل الانسجة.[5]
الأعراض
تختلف الأعراض في مرض تكون العظم الناقص من شخص إلى آخر، وكذلك بحسب نوع المرض، وتتراوح شدة المرض من الخفيف إلى المتوسط والشديد. فقد يعاني المريض من بضعة كسور فقط أو من عدد كبير من الكسور التي قد تشكل خطرا على حياته. ومن الأعراض العامة:
- ليونة في المفاصل.
- قصر في القامة.
- لون الصلبة يكون مائل للزرقة أو الرصاصي.
- إنحناء وتقوس العمود الفقري.
- ضعف في العضلات في بعض الانواع.
- زيادة في التعرق وعدم تحمل الحرارة.
- تقوس وإنحناء الساقين.
- رقَّة الجلد وسهوله ازرقاقه من الكدمات والضربات الصغيرة.
الأنواع وأعراضها
| النوع | الوصف | الجينة الوراثية | الأختصارات الوراثية للجينات (OMIM) | النمط الوراثي |
|---|---|---|---|---|
| I | اخف الأنواع حده والأكثر شيوعا | الكولاجين ألفا 1 | 166240 (IA), 166200 (IB) | جسمية مهيمنة، 60٪ دي نوفو [11] |
| II | أشد الأنواع حدة وتأثيراً | الكولاجين ألفا 1 والكولاجين ألفا 2 | 166210 (IIA) | جسمية مهيمنة، ~ 100٪ دي نوفو [11] |
| III | شديد مرفق بتشوهات | الكولاجين ألفا 1 والكولاجين ألفا 2 | 259420 | جسمية مهيمنة، ~ 100٪ دي نوفو [11] |
| IV | متوسط التأثير | الكولاجين ألفا 1 والكولاجين ألفا 2 | 166220 | جسمية مهيمنة، 60٪ دي نوفو novo [11] |
| V | يشترك مع النوع الرابع في المظاهر السريرية، يتميز بتكون نسيج العظم الكبير على شكل ("تشبه الغربال،") | مضاد الفيروسات التي يسببها بروتين عبر الغشاء5 | 610967 | جسمية مهيمنة [11][12] |
| VI | تشترك المظاهر السريرية نفسه من الرابع، ولكن ظاهرة فريدة من نوعها في تشكل الأنسيجية العظمية التي يطلق عليها ("نطاق الأسماك" | الصبغي(PEDF) المعروف أيضاً باسم بروتين السربين اف 1 | 610968 | مقهورة [11] |
| VII | يرتبط الغضروف بالبروتين | البروتين المرتبط بتكون جينات الغضروف | 610682 | مقهورة [11] |
| VIII | شديد وقاتل | بروتين ليبركان المرتبط بتكون العظم الناقص | 610915 | مقهورة |
يوجد ثمانية أنواع مكتشفة لمرض تكون العظم الناقص، وهي:
النوع الأول
يعتبر أخف الأنواع حدة وأكثرها شيوعاً، وقد لا يتم في أغلب الأحيان تشخيصه إلا بعد حدوث تكرار للكسور بشكل متتابع.
أعراضه: قد لا يكون هناك أي تشوهات في العظام، وتشمل اعراضه على:
- ضعف العضلات؛ وليونة المفاصل؛ تقوس خفيف جدا في العمود الفقري؛ تلون العين الصلبة.
- فقدان حاسة السمع في المراحل المبكرة عند بعض الأطفال.
- القدم المفلطحة ؛ التواء القدم والكسر ممكن حدوثه.
النوع الثاني
وهو أشد الأنواع حدة وتأثيرا.
أعراضه
| من سلسلة مقالات علم الأحياء |
| علم الوراثة |
|---|
 |
|
أساسيات مدخل كروموسوم |
|
مواضيع تعبير جيني الطبع مقابل التنشئة |
| بوابة علم الأحياء |
- الأطفال حديثين الولادة يولدون عاده بكسور متعددة؛ وجمجمة لينه وطريه.
- تشوهات شديدة في العظام وقصر القامة.
- مشاكل شديدة في الجهاز التنفسي ,ورئتان غير مكتملتا النمو مما يودي إلى وفاه المولود بعد ولادة مباشرة أو بعد الولادة بفترة قصيرة. وبسبب مشاكل التنفس يكونون بحاجه إلى الأكسجين بشكل دائم.
النوع الثالث
يتميز النوع الثالث بين الأنواع الأخرى بأنه يعتبر بأعراضه "تشوه تقدمي" نوع، اي انه يتعرض لحديثي الولادة مع أعراض خفيفة عند الولادة وتطور الأعراض في جميع مراحل الحياة. الأعمارفي هذا النوع قد تكون طبيعية، ولكن مع اعاقة بدنية شديدة.[13]
أعراضه
- تكسر العظام بشكل سهل جدا.
- يولدون بكسور متعددة وتبين الأشعة السينية التأم بعض الكسور أثناء مراحل نمو الجنين داخل رحم الأم.
- قصر القامة وإنحاء العمود الفقري وتشوهات شديدة في العظام الطويلة وفي بعض الأحيان القفص الصدري يكون بشكل البرميل.
- ضعف عضلات الذراعين والساقيين؛ مفاصل رخوة.
- تشكل الوجه الثلاثي.[14]
- تلون العين الصلبة.
- فقدان حاسة السمع في مراحل مبكرة.
- مشاكل في التنفس.
النوع الرابع
يعتبر متوسط الشدة. وتأثر المصاب مع هذا النوع يعتبر معتدل. ويمكن أن تتراوح شدة اعراضه في عدد قليل نسبيا من الكسور، كما هو الحال في النوع الأول، إلى شكل أكثر شدة تشبه النوع الثالث. سببه يكون في أن كمية الكولاجين قليلة وليست بما فيه الكفاية لتساعد في بناء العظم وتكوينه.
أعراضه
- سهوله تكسر العظام خاصة قبل سن البلوغ حيث تقل نسبه الكسور بعد سن البلوغ.
- المصابين بهذا النوع يكونون أقصر في القامة مقارنه بإفراد العائلة.
- تشوهات خفيفة إلى متوسطه في العظام.
النوع الخامس
يعتبر نوع معتدل. وهو مشابه في أعراضه للنوع الرابع من حيث تواتر الكسور ودرجة تشوه الهيكل العظمي. الميزة الأكثر وضوحاَ في هذا النوع هو، النسيج الضخامي الكبير في أكبر العظام الذي يتميز تشريحياً بشكل تكون فيه العظام "تشبه الغربال،".[13] سبب هذا النوع في بعض الحالات طفرة في بروتين مضاد الفيروسات عبر الغشاء 5 أف 5(IFITM5) الجينات.[12]
أعراضه
- تكلس في الغشاء بين العظمين بين اثنين من عظام الساعد مما يقيد دوران الساعد ويصعب تحويل المعصم ويمكن أن يسبب الخلع.
- احتمال إصابة النساء للكالس الضخامي في العظم الحرقفي.
- فقدان السمع.
- تشكل كمية كبيرة من بروتين إصلاح الأنسجة ( hyperplasic الكالس).
- خلع شعائي رئيسي ,(خلع شعائي كلي ).[15]
- تشكل العظام الطويلة.
النوع السادس
يعتبر نوع مقهور قاتل : تم تحديد نوعين من المتنحية التي تسبب هذا النوع، وسببه طفرة في بروتين الجين الغضروف المصاحب، والهيدروكسيلاز 1 المرتبط بالجين (LEPRE1).
أعراضه
- الام المصابة بهذا النوع لديها أحتمال 25٪ في إنجاب طفل مصاب به.
- الأشقاء غير المصابين بهذاحتما من 2 في ثلاثة نقل المرض لاطفالهم في المستقبل كون هذا النوع ينتقل عن طريق الجينات المتنحية.
النوع السابع
يعتبر نوع شديد إلى قاتل. لوحظ وجوده عام 2006 بين سكان الهنود الحمر القاطنين في كيبك في كندا.[16] الطفرات في الجين CRTAP والجين LEPRE1 يسبب هذا النوع.[17]
أعراضه
يصنف النوع السابع مثل النوع الثامن. [[17]]. وتشابه أعراضه في كثير من الجوانب أعراض النوع الرابع.
- ظهور الصلبة البيضاء عند الأطفال الرضع.
- تشكل رؤوس صغيرة، ووجوه مستديرة.
- عظام قصيرة في الساق، وعظم العضد، وعظم الفخذ.
- قصر القامة.
- ورك فحجاء، ويعتبر أمر شائع في هذا النوع.
النوع الثامن
يعتبر من الانواع الشديدة والقاتلة، وسببه ناتج عن غياب أو نقص حاد في نشاط بروتين Prolyl 3 هيدروكسيلاز بسبب طفرات في الجين LEPRE1.
أعراضه
تشبه أعراضه أعراض النوعين السادس والسابع إلا في حالة الصلبة البيضاء، وتتمثل الأعراض بشكل كبير في :.
- نقص حاد في مستويات النمو
- انحنائات وتقوصات في الهيكل العظمي.
جينات أخرى
اكتشاف أسرة مع المتنحيات التي قد تكون طفراتها أحد أسباب تشكل العظم الناقص ومنها: طفرات في الجينات على الكروموسوم 9.[18] يعتبر هذا الجين أحادي التكافؤ ومسؤول إطلاق الكالسيوم إلى داخل الخلايا.ومن المرجح حسب رأي الباحثين أنه يوجد جينات أخرى مازال البحث عنها جارياً.
التشخيص
عند التشخيص يتم ملاحظة الأعراض المرافقة ويتم أخذ عينة من الجلد لإجراء الفحوصات على الكولاجين، أيضاً تؤخذ عينة دم لإجراء الفحوصات والتحاليل على دي ان اي ( DNA). يتم تشخيص العظام في معظم الحالات عن طريق الأشعة السينية.
العلاج
لا يوجد علاج شافي لهذا المرض تماماً ولكن الهدف من العلاج هو زيادة قوة العظام وتجنب حدوث الكسور وزيادة الاعتماد على النفس في أمور ومتطلبات الحياة اليومية، فعند التعامل مع الشخص المصاب بتكون العظم الناقص يجب التركيز على قدرته الشخصية في إنجاز مهامه اليومية ومتطلباته وعلى نقاط الضعف والقوة التي يتميز بها بدلاً من التركيز على نوع تكون العظم الناقص المصاب به.و تتنوع طرق المعالجة إلى أنواع مختلفة مثل العلاج الجراحي، العلاج الدوائي والعلاج الطبيعي أو الوظيفي.
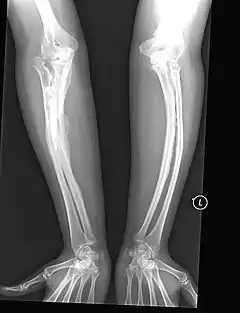
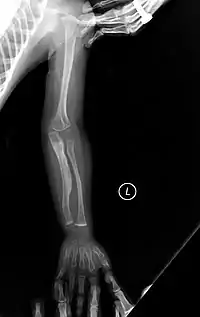
المعالجة الجراحية والإسعافية
- العناية بالكسور وتجبيرها.
- التدخل الجراحي عن طريق إدخال قضبان معدنية (أسياخ) في العظام الطويلة لتقليل تشوهات العظام والتحكم في الكسور، ولتحسين قوة العظم، وأول من وضع هذا الإجراء هو الطبيب هارولد سوفيلد (Harold A. Sofield)، في مستشفى شراينرز للأطفال في شيكاغو. خلال أواخر 1940.[19] وقد ثبت إجرائه هذا فعالية عالية في تقوية وتعزيز القنوات داخل النقي من العظام الطويلة. وكذلك في إعادة التأهيل والوقاية من الكسور؛ وبعدها اعتُمدت تجربته هذه في جميع أنحاء العالم.
كما أن إجراء الانصهار في العمود الفقري يمكن تصحيحه في الجنف، على الرغم من أن هشاشة العظام الكامنة تكون أكثر تعقيداً من هذه العملية. كما يمكن إجراء جراحة للظهور القاعدي ما إذا كانت الضغوط التي تمارس على الحبل الشوكي وجذع الدماغ هي التي تسبب المشاكل العصبية.
العلاج الطبيعي
العلاج الطبيعي يستخدم لتقوية العضلات وتحسين القدرة على الحركة بطريقة لطيفة، مع التقليل من خطر الإصابة بكسور. هذا غالباً ما ينطوي العلاج المائي واستخدام وسائد الدعم لتحسين الموقف. ويتم تشجيع الأفراد على تغيير المواقف بانتظام على مدار اليوم من أجل تحقيق التوازن بين العضلات التي تستخدم والعظام التي تتعرض لضغوط.
في حالة إصابة الأطفال فإنه غالباً مايكون هناك قلق كبير من محاولة نقلهم وتحريكهم نظراً لارتباطهم مع كل حركة بالألم. مما يجعل من الصعب إدارة العلاج الطبيعي لهم ويعقدها. إن استخدام المشاية، الكرسي المتحرك، المشدات وغيرها أشياء ضرورية ومهمة للاعتماد على النفس في قضاء أمور الحياة وزيادة الحركة. ففي السنوات الماضية كان آباء الأطفال المصابين بهذا المرض يتعاملون مع أبنائهم بحرص شديد عند التعامل على أبنائهم المصابين خصوصاً عند نقلهم من مكان إلى آخر أو في حملهم بالوسادة ولم يكن يُتوقع من الأطفال أن يقوموا بأي عمل ولو كان بسيطاً في الاعتماد على الذات مما أدى إلى معاملتهم كأطفال حديثي الولادة حتى في مراحل النمو والمراحل الدراسية. لكن في هذا الوقت أصبح العلاج الطبيعي والوظيفي الهدف البعيد المدى الذي يساعد الأشخاص المصابين في الاعتماد على أنفسهم في قضاء أمورهم الشخصية كالحياة الاجتماعية، العمل، الدراسة والتكيف مع وضعهم الصحي. إن العلاج الطبيعي والوظيفي مفيد في تقويه العضلات وتحسين الحركة والتقليل من أمكانيه التعرض للكسور. فتشجيع الأشخاص في تغير وضيعه جلوسهم وحركتهم من أجل تخفيف الضغط على منطقه معينه ومن ثم يخف الضغط على العضلات والعظام وهذا يؤدي إلى تقليل الإصابة بالكسور.
والعلاج الطبيعي والوظيفي مفيد في حالات أخرى مثل:
- الأطفال الذين لديهم تأخر في أداء المهارات الحركية.
- البرنامج التأهيلي بعد العملية الجراحيه أو كسر.
- الأشخاص الذين لديهم خوف من الحركة (بسبب تكرار الكسور ).
- الأشخاص الذين بحاجه لتعلم مهارة جديدة أو تعلم طريقه جديدة في إنجاز مهارة قد أنجزوها.
أثناء العمل مع الأشخاص المصابين وذويهم يجب على المعالج ملاحظة إن كان الشخص المصاب وأفراد عائلته هم من الأشخاص الذين يستطيعون التعايش مع المرض عندها يجب الاستماع إليهم وإلى نصائحهم من أجل الوصول إلى الهدف من العلاج الطبيعي والوظيفي، وأيضاً مع توفر مكان التدريب والأجهزة المتاحة فإن الأشخاص المصابين بالمرض يستطيعون العمل والتكيف مع ما هو متاح لهم في الأماكن التي يعيشون فيه بشكل أفضل.
العلاج الدوائي
لا تزال هناك دراسات قائمة على أدوية يتم أخذها بعد سن اليأس في التقليل من هشاشة العظام، لاستخدامه في حالات تكون العظم الناقص حيث أظهرت بعض الدراسات أن هذه الأدوية تزيد من كثافة فقرة العمود الفقري وتقلل من الآم العظام وبعض الدراسات أظهرت أنها تقلل من عدد مرات الكسور وتزيد من نسبه الحركة. الحالات الشديدة. في السنوات القليلة الماضية تم اكتشاف أدوية تسمى البايوفسونيت تساعد على التقليل من مضاعفات العظم الناقص. حيث أظهرت الدراسات أن هذا العلاج مفيد في التقليل من نسبه حدوث الكسور. ولكن استخدام الدواء على المدى الطويل قد يؤدي إلى تأخر شفاء الكسور. ويتم إعطاء الدواء عن طريق الفم أو عن طريق الوريد.[20][21][22] ويعتبرالبايفوسفونيت أقل فعالية لمعالجة تكون العظم الناقص حسب تقرير لمنظمة أوكسفام الدولية.[23] في عام 1998، أظهرت التجارب السريرية فعالية عقار باميدرونت (pamidronate ) في تقوية العظام وتقليل الإصابة بالكسور، والذي يُعطى في الوريد. وهو تطوير عن عقار فوسفونتس الذي كان يعطى سابقاً للبالغين في علاج ترقق العظام.[24] كماقد تعطى بعض المسكنات للألم وبعض مضادات الالتهاب.
التاريخ
يُتوقع أن اكتشاف مرض تكون العظم قديم جداً وتم تداوله على مر السنين وفي الأمم المختلفة. وكان يطلق عليه أسماء مثل متلازمة أيكمن-وبشتاين، متلازمة فروليك، ومرض تكون العظم الزجاجي. أما عن الاسم الحالي وهو اسم تكون العظم الناقص فيعود إلى عام 1895 [25]، وكان المصطلح الطبي المعتاد في القرن 20 حتى الوقت الحاضر.[26]
تم العثور على وصف قديم لحالة تكون العظم الناقص عند المصريين القدماء في حالة مومياء تعود إلى 1000 قبل الميلاد.[27] وحالة أخرى تعود إلى الملك الإسكندنافي ايفار الذي يقال أنه أُصيب بهذا المرض [28]
علم الأوبئة
تقول الإحصائيات التي جرت في الولايات المتحدة الأمريكية، إن معدل انتشار الإصابة بمرض العظم الناقص يكون في مولود واحد في كل 20,000 ولادة حية.[29][29] كما تعتبر دولة زيمبابوي من الدول ذات النسب العالية في الإصابة بالمرض وخاصة النوع الأول والثالث منه.[30]
مشاهير مصابون بالمرض

- الممثل الأمريكي مايكل أندرسون،.[31][32]
- ليو بيورمان. مخرج الأفلام الوثائقية القصيرة...[33]
- الممثلة البريطانية جولي فرناندز [34] Nabil Shaban,.[35]
- عازف الدرامز وعضو فريق (Toad the Wet Sprocket) الموسيقي راندي جوز.[36][37]
- فنان الهيب هوب كالين هيفرنان.[38]
- لاعب الألومبيات الأمريكي داوغ هيرلاند.[39]
- لاعب الهوكي الأمريكي تايلور ليبست.[40][41]
- انرجي مابوروتس عازف الماريمبا من زيمبابوي.[42][43]
- المغني والموسيقي الياباني يوشيكازو ميرا.[44]
- عازف الجاز الفرنسي الأصل أمريكي المولد مايكل بيتروسياني.[45]
- الكاتب والممثل الهندي الأصل البريطاني فردوس كانغا.[46]
- أقصر رجل في العالم الصيني هيي بينغينغ.[47]
- الشاعر والكاتب الألماني الجنسية المصري الأصل فيليب رحمة.[48]
- الرسام الفرنسي هنري تولوز لوترك.[49][50]
- قائد الفايكنغ ايافار راجنارسون الذي غزا بريطانيا.[51]
انظر أيضًا
 صور وملفات صوتية من كومنز
صور وملفات صوتية من كومنز
المراجع
- James, William; Berger, Timothy; Elston, Dirk (2005). Andrews' Diseases of the Skin: Clinical Dermatology. (10th ed.). Saunders. Page 517. ISBN 0-7216-2921-0.
- موسوعة الملك عبد الله بن عبد العزيز العربية للمحتوى الصحي [وصلة مكسورة] نسخة محفوظة 2020-09-13 على موقع واي باك مشين.
- Rauch F, Glorieux FH (2004). "Osteogenesis imperfecta". Lancet. 363 (9418): 1377–85. doi:10.1016/S0140-6736(04)16051-0. PMID 15110498. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Drögemüller, C.; Becker, D.; Brunner, A.; Haase, B.; Kircher, P.; Seeliger, F.; Fehr, M.; Baumann, U.; Lindblad-Toh, K.; Leeb, T. (2009). Barsh, Gregory S (المحرر). "A Missense Mutation in the SERPINH1 Gene in Dachshunds with Osteogenesis Imperfecta". PLoS Genetics. 5 (7): e1000579. doi:10.1371/journal.pgen.1000579. PMC 2708911. PMID 19629171. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Eckardt, J.; Kluth, S.; Dierks, C.; Philipp, U.; Distl, O. (2013). "Population screening for the mutation associated with osteogenesis imperfecta in dachshunds". Veterinary Record. 172 (14): 364. doi:10.1136/vr.101122. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Gautieri A, Uzel S, Vesentini S, Redaelli A, Buehler MJ (2009). "Molecular and mesoscale disease mechanisms of Osteogenesis Imperfecta". Biophysical Journal. 97 (3): 857–865. doi:10.1016/j.bpj.2009.04.059. PMC 2718154. PMID 19651044. الوسيط
|CitationClass=تم تجاهله (مساعدة)صيانة CS1: أسماء متعددة: قائمة المؤلفون (link) - What Is Osteogenesis Imperfecta? نسخة محفوظة 22 سبتمبر 2017 على موقع واي باك مشين.
- Osteogenesis Imperfecta in Children نسخة محفوظة 04 يوليو 2013 على موقع واي باك مشين.
- مصطلحات طبية تبدا بحرف تاء - صفحة 28 - ويب طب نسخة محفوظة 05 2يناير3 على موقع واي باك مشين.
- "Osteogenesis Imperfecta Foundation: Fast Facts". مؤرشف من الأصل في 09 أبريل 2019. اطلع عليه بتاريخ 05 يوليو 2007. الوسيط
|CitationClass=تم تجاهله (مساعدة); تحقق من التاريخ في:|تاريخ أرشيف=(مساعدة) - Steiner, RD (28 يناير 2005). "Osteogenesis Imperfecta". PMID 20301472. مؤرشف من الأصل في 17 أغسطس 2017. اطلع عليه بتاريخ 26 مارس 2012. الوسيط
|CitationClass=تم تجاهله (مساعدة); Cite journal requires|journal=(مساعدة) - Shapiro JR, Lietman C, Grover M, Lu JT, Nagamani SC, Dawson BC, Baldridge DM, Bainbridge MN, Cohn DH, Blazo M, Roberts TT, Brennen FS, Wu Y, Gibbs RA, Melvin P, Campeau PM, Lee BH (2013) Phenotypic variability of osteogenesis imperfecta type V caused by an IFITM5 mutation. J Bone Miner Res doi: 10.1002/jbmr.1891
- Glorieux FH, Rauch F, Plotkin H; et al. (2000). "Type V osteogenesis imperfecta: a new form of brittle bone disease". J. Bone Miner. Res. 15 (9): 1650–8. doi:10.1359/jbmr.2000.15.9.1650. PMID 10976985. الوسيط
|CitationClass=تم تجاهله (مساعدة); Explicit use of et al. in:|مؤلف=(مساعدة)صيانة CS1: أسماء متعددة: قائمة المؤلفون (link) - Page 771 in: Chen, Harold (2006). Atlas of genetic diagnosis and counseling. Totowa, NJ: Humana Press. ISBN 1-58829-681-4. مؤرشف من الأصل في 8 يونيو 2013. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Radial head dislocation | Radiology Reference Article | Radiopaedia.org نسخة محفوظة 21 أكتوبر 2017 على موقع واي باك مشين.
- "Recessive Form of OI Discovered by Foundation-funded Researcher" (PDF). مؤرشف من الأصل (PDF) في 03 مارس 2016. الوسيط
|CitationClass=تم تجاهله (مساعدة); تحقق من التاريخ في:|تاريخ أرشيف=(مساعدة) - Genetics Home Reference: Genetic Conditions>
Osteogenesis imperfecta (Reviewed November 2007) "نسخة مؤرشفة". Archived from the original on 29 أبريل 2010. اطلع عليه بتاريخ 12 أغسطس 2013. الوسيط
|CitationClass=تم تجاهله (مساعدة)صيانة CS1: BOT: original-url status unknown (link) - Volodarsky M, Markus B, Cohen I, Staretz-Chacham O, Flusser H, Landau D, Shelef I, Langer Y, Birk OS (2013) A deletion mutation in TMEM38B associated with autosomal recessive osteogenesis imperfecta. Hum Mutat doi: 10.1002/humu.22274
- "Chicago Shriners Hospital - Osteogenesis imperfecta". مؤرشف من الأصل في 28 سبتمبر 2007. اطلع عليه بتاريخ 05 يوليو 2007. الوسيط
|CitationClass=تم تجاهله (مساعدة) - DiMeglio LA, Peacock M (2006). "Two-year clinical trial of oral alendronate versus intravenous pamidronate in children with osteogenesis imperfecta". J. Bone Miner. Res. 21 (1): 132–40. doi:10.1359/JBMR.051006. PMID 16355282. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Risedronate in children with osteogenesis imperfecta: a randomised, double-blind, placebo-controlled trial, Lancet, 6 August 2013 نسخة محفوظة 04 نوفمبر 2013 على موقع واي باك مشين.
- Oral bisphosphonates for paediatric osteogenesis imperfecta? Lancet August 6, 2013 نسخة محفوظة 02 نوفمبر 2013 على موقع واي باك مشين.
- Chevrel G, Schott AM, Fontanges E; et al. (2006). "Effects of oral alendronate on BMD in adult patients with osteogenesis imperfecta: a 3-year randomized placebo-controlled trial". J. Bone Miner. Res. 21 (2): 300–6. doi:10.1359/JBMR.051015. PMID 16418786. الوسيط
|CitationClass=تم تجاهله (مساعدة); Explicit use of et al. in:|مؤلف=(مساعدة)صيانة CS1: أسماء متعددة: قائمة المؤلفون (link) - Glorieux FH, Bishop NJ, Plotkin H, Chabot G, Lanoue G, Travers R (1998). "Cyclic administration of pamidronate in children with severe osteogenesis imperfecta". N. Engl. J. Med. 339 (14): 947–52. doi:10.1056/NEJM199810013391402. PMID 9753709. الوسيط
|CitationClass=تم تجاهله (مساعدة)صيانة CS1: أسماء متعددة: قائمة المؤلفون (link)Free full text - K. Buday, Beiträge zur Lehre von der Osteogenesis imperfecta (1895)
- Sillence DO, Senn A, Danks DM (1979). "Genetic heterogeneity in osteogenesis imperfecta". J. Med. Genet. 16 (2): 101–16. doi:10.1136/jmg.16.2.101. PMC 1012733. PMID 458828. الوسيط
|CitationClass=تم تجاهله (مساعدة)صيانة CS1: أسماء متعددة: قائمة المؤلفون (link) - "Osteogenesis Imperfecta Foundation: Glossary". مؤرشف من الأصل في 13 يونيو 2018. اطلع عليه بتاريخ 05 يوليو 2007. الوسيط
|CitationClass=تم تجاهله (مساعدة) - synd/1743 على قاموس من سمى هذا؟
- eMedicine> Osteogenesis Imperfecta Author: Horacio Plotkin. Updated: March 2, 2010 نسخة محفوظة 07 نوفمبر 2017 على موقع واي باك مشين.
- Viljoen D, Beighton P (1987). "Osteogenesis imperfecta type III: an ancient mutation in Africa?". Am. J. Med. Genet. 27 (4): 907–12. doi:10.1002/ajmg.1320270417. PMID 3425600. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Cast: Michael J. Anderson". Mulholland-drive.net. مؤرشف من الأصل في 04 مارس 2016. اطلع عليه بتاريخ 15 أغسطس 2011. الوسيط
|CitationClass=تم تجاهله (مساعدة); تحقق من التاريخ في:|تاريخ أرشيف=(مساعدة) - Dargis, Manohla (2005-02-07). "We're Sorry". The New York Times. مؤرشف من الأصل في 05 فبراير 2016. اطلع عليه بتاريخ 20 مايو 2010. الوسيط
|CitationClass=تم تجاهله (مساعدة); تحقق من التاريخ في:|تاريخ أرشيف=(مساعدة) - LJ World نسخة محفوظة 09 نوفمبر 2013 على موقع واي باك مشين. [وصلة مكسورة]
- "Julie Fernandez - founder of The Disability Foundation". Tdf.org.uk. مؤرشف من الأصل في 23 مايو 2013. اطلع عليه بتاريخ 15 أغسطس 2011. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Banks-Smith, Nancy (2003-06-13). "Short and to the point". The Guardian. London. مؤرشف من الأصل في 23 فبراير 2006. اطلع عليه بتاريخ 20 مايو 2010. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "randy+guss"+osteogenesis&hl=en&gl=us&pid=bl&srcid=ADGEEShZCs41L1L-ar3Al70XOCbnjF9quVJACAMkhVI8ve6-YsU_8BJ71Tf82kgJhElr58jANqasUneJS0zvDBQAR6iD6cTJAbQ_JKIpr-wiHjSvtPpRmEvkZa4wNhfEAB3S_eQ4EpLH&sig=AHIEtbR-mcu_iTenwddNRTiy4qXs3sTXmQ "OIF Newsletter" (PDF). Docs.google.com. مؤرشف من "randy+guss"+osteogenesis&hl=en&gl=us&pid=bl&srcid=ADGEEShZCs41L1L-ar3Al70XOCbnjF9quVJACAMkhVI8ve6-YsU_8BJ71Tf82kgJhElr58jANqasUneJS0zvDBQAR6iD6cTJAbQ_JKIpr-wiHjSvtPpRmEvkZa4wNhfEAB3S_eQ4EpLH&sig=AHIEtbR-mcu_iTenwddNRTiy4qXs3sTXmQ الأصل (PDF) في 09 أبريل 2016. اطلع عليه بتاريخ 15 أغسطس 2011. الوسيط
|CitationClass=تم تجاهله (مساعدة); تحقق من التاريخ في:|تاريخ أرشيف=(مساعدة) - Ragogna, Mike (2010-06-15). "Huffington Post". Huffington Post. مؤرشف من الأصل في 29 مايو 2016. اطلع عليه بتاريخ 15 أغسطس 2011. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Wheelchair Sports Camp's Crip Life". مؤرشف من الأصل في 7 أبريل 2015. اطلع عليه بتاريخ أغسطس 2020. الوسيط
|CitationClass=تم تجاهله (مساعدة); تحقق من التاريخ في:|تاريخ الوصول=(مساعدة) - Tone. "Rowing at Pacific Lutheran University began in 1964" (PDF). Google.com. مؤرشف من الأصل (PDF) في 7 مايو 2020. اطلع عليه بتاريخ 15 أغسطس 2011. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "USA Hockey" (PDF). webcache.googleusercontent.com. مؤرشف من الأصل (PDF) في 8 مايو 2020. اطلع عليه بتاريخ 15 أغسطس 2011. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Paralympics info, McKinney Courier-Gazette, 6 April 2006. [وصلة مكسورة] نسخة محفوظة 19 فبراير 2020 على موقع واي باك مشين.
- "Bergen County Record". Highbeam.com. مؤرشف من الأصل في 24 سبتمبر 2015. اطلع عليه بتاريخ 15 أغسطس 2011. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Rutgers". News.rutgers.edu. 2008-12-23. مؤرشف من الأصل في 05 يونيو 2013. اطلع عليه بتاريخ 15 أغسطس 2011. الوسيط
|CitationClass=تم تجاهله (مساعدة); تحقق من التاريخ في:|تاريخ أرشيف=(مساعدة) - Biography with images at bach-cantatas.com نسخة محفوظة 13 يوليو 2017 على موقع واي باك مشين.
- Michel Petrucciani: Victory of the Spirit, International Herald Tribune, 12 January 1999. نسخة محفوظة 26 أبريل 2020 على موقع واي باك مشين.
- Dallas - Movies - Willing and able[وصلة مكسورة] نسخة محفوظة 09 سبتمبر 2009 على موقع واي باك مشين.
- The Smallest Man of the World is Dead, Explore Everything, 26 April 2010. [وصلة مكسورة] نسخة محفوظة 07 2يناير0 على موقع واي باك مشين.
- CBI.gov نسخة محفوظة 09 2يناير5 على موقع واي باك مشين.
- "Henri de Toulouse-Lautrec> Biography> Chronology". Toulouselautrec.free.fr. مؤرشف من الأصل في 16 مايو 2013. اطلع عليه بتاريخ 15 أغسطس 2011. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Noble figure". The Guardian. London. 2004-11-20. مؤرشف من الأصل في 25 يونيو 2008. اطلع عليه بتاريخ 20 مايو 2010. الوسيط
|CitationClass=تم تجاهله (مساعدة) - The Vikings, Frank. R. Donovan, author; Sir Thomas D. Kendrick, consultant; Horizon Caravel Books, by the editors of Horizon Magazine, Fourth Edition, American Heritage Publishing Co.: New York, 1964, LCC# 64-17106, pp. 44-45; 145, 148.
 بوابة طب
بوابة طب