خلل التقرن الخلقي
خلل التقرن الخلقي (بالإنجليزية: Dyskeratosis congenita)[3] (باختصار DKC) هو اضطراب خلقي نادر، يكون النمط الظاهري له متغير للغاية.[4]تم تعريف هذا المرض بشكل كلاسيكي وذلك عند ظهور تصبغ الجلد غير الطبيعي، وحثل الأظلافر،[5] وصداف الغشاء المخاطي الفمي، ولكن هذه الأعراض لا تحدث دائمًا. يتميز هذا المرض بقسيمات طرفية قصيرة. بعض مظاهر هذا المرض تشبه الشيخوخة المبكرة.
| خلل التقرن الخلقي | |
|---|---|
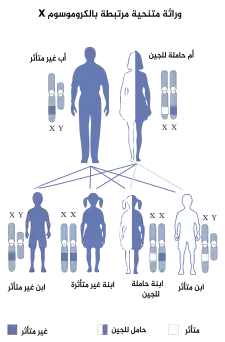 خلل التقرن الخلقي هو اضراب يتم وراثته بواسطة الوراثة المتنحية المرتبطة بX. خلل التقرن الخلقي هو اضراب يتم وراثته بواسطة الوراثة المتنحية المرتبطة بX. | |
| تسميات أخرى | Zinsser-Cole-Engman syndrome,[1][2]:570 |
| معلومات عامة | |
| الاختصاص | علم الوراثة الطبية |
| من أنواع | مرض جلدي ، وعسر التقرن |
يؤثر المرض في البداية بشكل أساسي على الجلد، ولكن أحد النتائج الرئيسية لذلك هو الفشل التدريجي لنخاع العظام الذي يحدث في أكثر من 80% من الحالات، مما يتسبب في الوفاة المبكرة.[4]
الانتشار الدقيق لخلل التقرن الخلقي غير معروف. تشير التقديرات إلى أن يحدث في حوالي 1 من كل مليون شخص.[6]
الظهور
يتسم خلل التقرن الخلقي بوجود تصبغ جلدي، والشيب المبكر، وحثل الأظلافر، وصداف الغشاء المخاطي الفمي، والتمزق المستمر بسبب رتق القناة الدمعية، وغالباً نقص الصفيحات، وفقر الدم، وضمور الخصية في الذكور الحاملة للمرض، والعرضة للسرطان.
العديد من هذه الأعراض هي سمة من سمات أمراض الشيخوخة، وغالباً ما تؤدي الأشكال الأكثر خطورة من هذا المرض إلى الوفاة المبكرة لمن يحملها.
الأسباب
في حوالي نصف الأشخاص الذين يعانون من خلل التقرن الخلقي، يحدث الاضطراب بسبب طفرات في جين TERT أو TERC أو DKC1 أو TINF2. توفر هذه الجينات تعليمات لصنع البروتينات التي تساعد في الحفاظ على الهياكل المعروفة باسم التيلوميرات، والتي توجد في نهايات الكروموسومات. تلعب التيلوميرات دوراً أساسياً في الحفاظ على الكروموسومات أثناء انقسام الخلية، مما يضمن نجاح عملية الانقسام الخلوي. في هذا المرض، تُسبب هذه الطفرات خلل في البروتينات المسؤولة عن صيانة التيلوميرات، وبالتالي هذا يتسبب بمشاكل في صيانة التيلوميرات، مما يؤدي إلى موت الخلايا بسرعة عند انقسامها. (خاصة الخلايا التي تنقسم بشكل دائم، مثل خلايا الجلد والأظافر).
في عدد صغير من الأفراد المصابين بخلل التقرن الخلقي، تم تحديد طفرات في جينات أخرى معنية بصيانة التيلومير. الأفراد المصابون الآخرون ليس لديهم طفرات في أي من الجينات المرتبطة حاليًا بمرض خلل التقرن الخلقي. في هذه الحالات، سبب الاضطراب غير معروف، ولكن من المحتمل أن تكون هناك جينات مجهولة أخرى مرتبطة بصيانة التيلومير.[6]
تساعد التيلوميرات في حماية الكروموسومات من الالتصاق أو التحلل بشكل غير طبيعي. في معظم الخلايا، تصبح التيلوميرات أقصر تدريجياً عندما تنقسم الخلية. بعد عدد معين من الانقسامات الخلوية، تصبح التيلوميرات قصيرة للغاية بحيث تؤدي إلى إيقاف الخلية عن الانقسام أو التدمير الذاتي (تخضع للاستماتة).[6]
يتم الحفاظ على التيلومير بواسطة معقدين بروتينيين مهمين يطلق عليهما التيلوميراز والشيلتيرن (يُسمى أيضًا التيلوزوم).[7] يساعد التيلوميراز في الحفاظ على طول التيلومير الطبيعي عن طريق إضافة شرائح صغيرة متكررة من الحمض النووي إلى نهايات الكروموسومات في كل مرة تنقسم فيها الخلية. يتم إنتاج المكونات الرئيسية للتيلوميراز، التي تسمى hTR و hTERT، من جينات TERC و TERT، على التوالي. المكون hTR هو جزيء الرنا، "ابن عم" كيميائي للدنا. يوفر ذلك قالبًا لإنشاء تسلسل متكرر للحمض النووي الذي يضيفه التيلوميراز إلى نهايات الكروموسومات.[6]
تتمثل وظيفة المكون hTERT في إضافة شريحة DNA الجديدة إلى نهايات الكروموسوم. يوفر جين DKC1 تعليمات لصنع بروتين آخر مهم في وظيفة التيلوميراز. هذا البروتين، المسمى dyskerin، يرتبط بـ hTR ويساعد على تثبيت معقد التيلوميراز.[6]
في المقابل، يساعد معقد الشيلتين على حماية التيلوميرات من عملية إصلاح الحمض النووي للخلية. بدون حماية الشيلتين، ستشعر آلية الإصلاح بأن الكروموسوم ينتهي ككسر غير طبيعي في تسلسل الحمض النووي وإما أن تحاول تلك الآلية ضم النهايات معًا أو الشروع في عملية الاستماتة. يوفر جين TINF2 تعليمات لصنع بروتين يمثل جزءًا من معقد الشيلتين.[6]
تؤدي الطفرات في الجينات: TERT أو TERC أو DKC1 أو TINF2 إلى خلل في معقدات التيلوميراز أو الشيلتين، مما يؤدي إلى ضعف صيانة التيلوميرات وانخفاض طولها. الخلايا التي تنقسم بسرعة معرضة بشكل خاص لآثار انخفاض طول التيلوميرات. نتيجة لذلك، قد يعاني الأشخاص الذين يعانون من خلل التقرن الخلقي مجموعة متنوعة من المشكلات التي تؤثر على الخلايا السريعة الانقسام في الجسم مثل خلايا فراش الظفر، وبصيلات الشعر، والجلد، وبطانة الفم (الغشاء المخاطي للفم)، ونخاع العظام.[6]
قد يؤدي انكسار وعدم استقرار الصبغيات الناتجة عن عدم كفاية صيانة التيلومير إلى تغييرات وراثية تسمح للخلايا بالانقسام بطريقة غير خاضعة للمراقبة، مما يؤدي إلى تطور السرطان لدى الأشخاص المصابين بخلل التقرن الخلقي.[6]
العرضة للسرطان
للوهلة الأولى، تبدو القابلية للإصابة بالسرطان غير بديهية لأن إعادة تنشيط التيلوميراز في العديد من السرطانات المعروفة هي في الواقع خطوة مطلوبة لتطور السرطان والخباثة (طالع صفحة التيلومير). في أي مرض يتأثر فيه التيلوميراز، لا يبدو أنه يتبع أن السرطان سيكون من المضاعفات الناجمة عنه. يلاحظ المؤلفون الطبيعة المتناقضة للإصابة بالسرطان لدى الأفراد الذين يبدو أنهم يفتقرون إلى أحد المكونات المطلوبة للسرطان.
يِعتقد أنه[8]بدون التيلوميراز الوظيفية، يكون من المحتمل أن يتم ربط الكروموسومات معًا عند نهاياتها من خلال طريق الربط غير المتماثل. هذا يثبت أنه أمر شائع بما فيه الكفاية، والخباثة حتى من دون وجود التيلوميراز ممكنة.
الفزيولوجيا المرضية
خلل التقرن الخلقي هو اضطراب في ضعف صيانة التيلومير. ويرجع ذلك أساسًا إلى عدد من الطفرات الجينية التي تؤدي إلى وظيفة الريبوسوم غير الطبيعية[8]، والتي تسمى اعتلال الريبوسوم (Ribosomopathy). على وجه التحديد، يرتبط المرض بواحد أو أكثر من الطفرات التي تؤثر بشكل مباشر أو غير مباشر على مكون الحمض النووي الريبي التيلوميراز (TERC).
التيلوميراز هو منتسخة عكسية تحافظ على تسلسل تكرار محدد للحمض النووي، التيلومير، أثناء التطور. يتم وضع التيلوميرات بواسطة التيلوميراز على طرفي الكروموسومات الخطية كوسيلة لحماية الحمض النووي الخطي من الأشكال العامة للتلف الكيميائي وتصحيح تقصير نهاية الكروموسومات الذي يحدث أثناء تضاعف الحمض النووي الطبيعي.[9]
التشخيص
قد يشتبه في تشخيص خلل التقرن الخلقي بناءً على تقييم سريري شامل وتاريخ المريض المفصل وتحديد النتائج المميزة خاصة التغيرات في الجلد أو الفم.[10] في الأفراد الذين يصابون بفقر الدم اللاتنسجي كعلامة أولى للاضطراب أو تشخيص التليف الرئوي يكون أكثر صعوبة.[10]
قد تدعم التيلوميرات القصيرة جدًا في خلايا الدم المحيطية تشخيص خلل التقرن لدى المرضى الذين يعانون من فشل النخاع العظمي.[10]
يمكن أن تؤكد الاختبارات الوراثية الجزيئية لتحديد الطفرات في جين DKC1 أو TERC أو TERT أو TINF2 NHP2 أو NOP10 تشخيص خلل التقرن الخلقي. ومع ذلك، فإن الاختبارات الجينية الإكلينيكية تكون غالباً باهظة الثمن، ولا تتاح إلا لبعض البحوث على أساس جيني.[10]
علاوة على ذلك، لا يكتشف الفحص الجيني عادةً عمليات الحذف الجينية الكبيرة، وبالتالي فإن المرضى الذين يعانون من المرض بسبب الحذف الجيني الكبير لا يمكن عادةً تشخيصهم. قد يكون من الصعب أيضًا إثبات أن متغير التسلسل المحدد هو في الواقع مسؤول عن المرض. ليست كل الطفرات المُسجلة في المصادر الطبية مسؤولة في الواقع عن المرض (الأشكال المتعددة) أو تسبب المرض في جميع الأفراد (الاختراق المتغير). في حوالي 50% من المرضى، لم يتم تحديد أي طفرة، على الرغم من وجود الأعراض السريرية الكلاسيكية.[10]
المآل
يرتبط المرض عادةً بمتوسط عمر أقصر، لكن الكثيرين يعيشون حتى سن 60 على الأقل.[11]
الأسباب الرئيسية لخطورة المرض تشمل BMF، والسرطان والمضاعفات الرئوية.[12]يتراوح العمر المتوقع من الطفولة إلى فترة طويلة في العقد السابع. ما يصل إلى 40% من المرضى يكون لديهم BMF بحلول سن 40.[12]
العلاج
لا يوجد أي إجماع حول كيفية علاج المرضى الذين يعانون من خلل التقرن الخلقي. المصادر الطبية متحيزة تجاه العلاج ونتائج العلاج للمرضى الذين يقدمون مع الشكل الكلاسيكي للمرض. لا يزال هناك القليل المعروف عن العلاج ومراقبة الأمراض من الأفراد الذين يعانون من مرض غير نمطي أو صامت.[10]
يتم توجيه علاج خلل التقرن الخلقي نحو الأعراض المحددة الظاهرة لدى كل فرد. قد يتطلب العلاج جهود منسقة لفريق من المتخصصين. قد يحتاج أطباء الأطفال وأخصائيي الأمراض الباطنة وأخصائي أمراض الدم وأخصائيي الأمراض الجلدية وأخصائيي الوراثة الطبية وأخصائيي السرطان (أطباء الأورام) وغيرهم من المتخصصين في الرعاية الصحية إلى التخطيط بشكل منهجي وشامل لعلاج الشخص المصاب.[10]
تشمل توصيات العلاج العامة للأفراد المصابين بخلل التقرن الخلقي تجنب التدخين والكحول للحفاظ على الرئتين والكبد واستخدام الكريمات المرطبة لمنع تلف الجلد.[10]
قد تساعد النظافة الجيدة للأسنان على منع فقدان الأسنان مبكرًا وتؤخر تطور ورم خبيث في اللسان. في بعض المرضى، يستجيب فشل النخاع العظمي ونقص المناعة بشكل عابر للأندروجينات وهرمونات النمو المكونة للدم.[10]
الأندروجينات (على سبيل المثال، أوكسي ميثولون)، والتي هي نوع من الهرمونات الذكرية المصطنعة، قد تزيد من خلايا الدم الحمراء، وفي كثير من الأحيان إنتاج الصفائح الدموية في بعض الأفراد. يمكن استكمال علاج الأندروجين بالعلاج بالكورتيكوستيرويد (على سبيل المثال، بريدنيزون)، مما قد يؤخر تسارع النمو المحتمل أن يرتبط بعلاج الأندروجين ويقلل النزيف المرتبط بنقص الصفيحات.[10]
تم أيضاً استخدام فئة من العقاقير تعرف باسم عوامل نمو المكونة للدم لعلاج الأفراد الذين يعانون من خلل التقرن الخلقي، وخاصةً عامل تحفيز مستعمرة المحبب (G-CSF) وعامل تحفيز مستعمرة المحببة (GM-CSF). قد تؤدي عوامل النمو هذه إلى زيادة إنتاج بعض خلايا الدم البيضاء (العدلات) بشكل عابر. في حالات نادرة، يزيد العلاج بهذه الأدوية من مستويات خلايا الدم الحمراء والصفائح الدموية.
في معظم الحالات، تكون فوائد الأندروجينات وعوامل النمو مؤقتة فقط. يختلف مقدار الوقت المحدد لهذه العلاجات في تحسين وظيفة النخاع العظمي في كل حالة على حدة.[10]
إذا كان من الممكن العثور على متبرع متوافق، يمكن أن تؤدي عملية زرع الخلايا الجذعية المكونة للدم إلى علاج تشوهات الدم المرتبطة بخلل التقرن الخلقي. وينبغي النظر خصوصاً في زرع الخلايا الجذعية المكونة للدم في المرضى الذين يعانون من فشل النخاع العظمي بشكل رئيسي.[10]
زرع الخلايا الجذعية المكونة للدم لا يحسن الأنسجة المتضررة من خلل التقرن الوراثي. المرضى الذين يعانون من خلل التقرن الخلقي لديهم حساسية متزايدة تجاه الإشعاع وبعض أدوية العلاج الكيميائي. فوج التكييف البديل هو بدون تشعيع أو يجب تجنب بعض أدوية العلاج الكيميائي مثل بوسلفان أو الملفان. المضاعفات الرئوية بعد زرع الخلايا الجذعية المكونة للدم ليست غير شائعة وقد تكون قاتلة في العديد من الأحيان.[10]
فرط الحساسية للأفراد الذين يعانون من خلل التقرن الخلقي للإشعاع والعلاج الكيميائي يعيق علاج السرطان لدى هؤلاء الأفراد. الاستئصال الجراحي للسرطان هو على الأرجح السطر الأول من العلاج.
يجب مراقبة الأفراد المصابين بتطور أمراض الرئة والسرطان. قد تكون الاستشارة الوراثية ذات فائدة للأفراد المصابين وأسرهم. العلاجات الأخرى هي إما عرضية و/أو داعمة و/أو تلطيفية.
الأبحاث
استخدمت الأبحاث الحديثة الخلايا الجذعية المحفزة المستحثة لدراسة آليات المرض لدى البشر، واكتشفت أن إعادة برمجة الخلايا الجسدية يعيد استطالة التيلومير في خلايا خلل التقرن الخلقي (DKC) على الرغم من الآفات الجينية التي تؤثر على التيلوميراز، والذي بدوره يؤثر على التيلومير. كانت خلايا DKC المعاد برمجتها قادرة على التغلب على قيود حرجة في مستويات TERC والوظائف المستعادة (صيانة التيلومير والتجديد الذاتي). علاجياً، يمكن أن تكون الأساليب التي تهدف إلى زيادة التعبير عن TERC مفيدة في DKC.[13]
الوبائيات
الانتشار الدقيق لخلل التقرن الخلقي غير معروف، حيث يصعب تقييم مدى انتشار أو حدوث خلل التقرن الخلقي. تشير التقديرات إلى أن أنه يحدث في حوالي 1 من مليون شخص.[14]
في عدد السكان الذين يعانون من فشل النخاع العظمي حوالي 2-5% من المرضى يعانون من هذا المرض بسبب خلل التقرن الخلقي. في المرضى الذين يعانون من التليف الرئوي، يُعتقد أن 2-5% ناتج عن طفرات في TERC أو TERT.[10]
في العائلات التي تتزايد فيها نسبة فشل النخاع العظمي و/أو أمراض الرئة، ينبغي استبعاد خلل التقرن الخلقي كسبب محتمل لمرضها.[10]تم الإبلاغ عن أكثر من 400 أسرة في العالم.[12]
اضرابات متعلقة
يمكن أن تكون أعراض الاضطرابات التالية مماثلة لأعراض خلل التقرن الخلقي. قد تكون المقارنات مفيدة للتشخيص التفريقي.[15]
- أنيميا فانكوني، المعروف أيضاً باسم فقر الدم اللاتنسجي مع بعض الشذوذات الخلقية، هو اضطراب وراثي نادر قد يكون واضحاً عند الولادة أو أثناء الطفولة. في بعض الحالات، قد لا يتم تشخيص أنيميا فانكوني حتى مرحلة البلوغ. يتم تعريفه على أنه العرضة الموروثة لطفرات الجينات، ربما بسبب ضعف القدرة على إصلاح أضرار الكروموسوم (عدم الاستقرار الصبغي). يتميز هذا الاضطراب بنقص في جميع عناصر نخاع العظام بما في ذلك خلايا الدم الحمراء وخلايا الدم البيضاء والصفائح الدموية (قلة الكريات الشاملة). قد يرتبط أنيميا الفانكوني أيضًا بالقلب والكلى و/أو تشوهات الهيكل العظمي. عادة ما يكون مصحوبًا بتغير لونه غير مكتمل اللون (تغير تصبغ) الجلد. هناك عدة أنواع فرعية مختلفة (مجموعات مكملة) لأنيميا فانكوني، يعتقد أن كل منها ينتج عن تغيير غير طبيعي (طفرة) في جين مختلف. يبدو أن كل نوع فرعي يتشارك في نفس الأعراض والنتائج المميزة (النمط الظاهري). معظم حالات أنيميا فانكوني لها وراثة جسمية متنحية. لا يرتبط أنيميا فانكوني بأي شكل من الأشكال بمتلازمة فانكوني، وهو اضطراب نادر في الكلى.[15]
- فقر الدم اللاتنسجي المكتسب هو اضطراب نادر ناتج عن فشل عميق في نخاع العظم. النخاع العظمي هو المادة الإسفنجية الموجودة في وسط العظام الطويلة للجسم. ينتج النخاع العظمي خلايا متخصصة (الخلايا الجذعية المكونة للدم) التي تنمو وتتطور في نهاية المطاف إلى خلايا الدم الحمراء وخلايا الدم البيضاء والصفائح الدموية. في فقر الدم اللاتنسجي المكتسب، يؤدي الغياب شبه الكامل للخلايا الجذعية المكونة للدم في النهاية إلى انخفاض مستويات خلايا الدم الحمراء والبيضاء والصفائح الدموية (قلة الكريات الشاملة). قد تختلف الأعراض المحددة المرتبطة بفقر الدم اللاتنسجي المكتسب، ولكنها تشمل التعب والإلتهابات المزمنة والدوخة والضعف والصداع وحلقات النزيف المفرط. في بعض الحالات يكون فقر الدم اللاتنسجي المكتسب ثانويًا للاضطرابات الأخرى أو التعرض (على سبيل المثال بعض الأدوية أو التعرض للسموم أو الإشعاع). ومع ذلك، في معظم الحالات، سبب تطور فقر الدم اللاتنسجي المكتسب غير معروف. يصنف هؤلاء المرضى عادةً على أنهم مصابون ب "فقر الدم اللاتنسجي مجهول السبب".[15]
فضلاً عن ذلك، إن الاختبارات الجينية لدى مرضى فقر الدم اللاتنسجي مجهول السبب تظهر بشكل متزايد أن بعض هؤلاء المرضى لديهم بالفعل مرض وراثي أو لديهم استعداد وراثي لتطوير فقر الدم اللاتنسجي. يعاني حوالي 2-5% من المرضى الذين تم تشخيصهم في الأصل من فقر الدم اللاتنسجي مجهول السبب (وبعض الحالات مع خلل التنسج النقوي، في الواقع من خلل وراثي مثل مرضى خلل التقرن الخلقي، لكن في وقت التشخيص كانوا يفتقرون إلى السمات المميزة الأخرى لخلل التنسج الخلقي)، أو كانت هذه معتدلة لدرجة أنه ربما لم يتم التعرف عليها. من المثير للجدل ما إذا كان ينبغي تصنيف المرض لدى المرضى على أنه "خلل التقرن الخلقي" أو "خلل التقرن الشاذ" أو "فقر الدم اللاتنسجي مع التيلوميرات القصيرة". يبدو أن الجهاز المناعي للمريض يلعب دورًا في استمرار العديد من حالات فقر الدم اللاتنسجي. يعتقد بعض الباحثين أن الجهاز المناعي هو السبب الرئيسي للعديد من حالات فقر الدم اللاتنسجي مجهول السبب. يعتمد هذا على استجابة نصف المرضى تقريبًا للتثبيط المناعي، على سبيل المثال ضد جلوبيولين الغدة الصعترية (ATG) أو السيكلوسبورين أو السيكلوفوسفاميد.[15]
المراجع
- الوراثة المندلية البشرية عبر الإنترنت (OMIM) 305000
- James, William; Berger, Timothy; Elston, Dirk (2005). Andrews' Diseases of the Skin: Clinical Dermatology. (10th ed.). Saunders. (ردمك 0-7216-2921-0).
- Team, Almaany. "ترجمة و معنى dyskeratosis congenita بالعربي في قاموس المعاني. قاموس عربي انجليزي مصطلحات صفحة 1". www.almaany.com (باللغة الإنجليزية). مؤرشف من الأصل في 9 يوليو 2019. اطلع عليه بتاريخ 09 يوليو 2019. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "OMIM Entry - # 127550 - DYSKERATOSIS CONGENITA, AUTOSOMAL DOMINANT 1; DKCA1". omim.org. مؤرشف من الأصل في 4 يوليو 2017. اطلع عليه بتاريخ 09 يوليو 2019. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Team, Almaany. "ترجمة و معنى nail dystrophy بالعربي في قاموس المعاني. قاموس عربي انجليزي مصطلحات صفحة 1". www.almaany.com (باللغة الإنجليزية). مؤرشف من الأصل في 9 يوليو 2019. اطلع عليه بتاريخ 09 يوليو 2019. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Reference, Genetics Home. "Dyskeratosis congenita". Genetics Home Reference (باللغة الإنجليزية). مؤرشف من الأصل في 7 فبراير 2019. اطلع عليه بتاريخ 09 يوليو 2019. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Xin, Huawei; Liu, Dan; Songyang, Zhou (2008). "The telosome/shelterin complex and its functions". Genome Biology. 9 (9): 232. doi:10.1186/gb-2008-9-9-232. ISSN 1465-6906. PMID 18828880. مؤرشف من الأصل في 17 يناير 2021. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Vulliamy, Tom; Marrone, Anna; Goldman, Frederick; Dearlove, Andrew; Bessler, Monica; Mason, Philip J.; Dokal, Inderjeet (2001-09). "The RNA component of telomerase is mutated in autosomal dominant dyskeratosis congenita". Nature. 413 (6854): 432–435. doi:10.1038/35096585. ISSN 0028-0836. مؤرشف من الأصل في 02 ديسمبر 2008. الوسيط
|CitationClass=تم تجاهله (مساعدة); تحقق من التاريخ في:|تاريخ=(مساعدة) - Greider, Carol W. (1996-06-01). "Telomere length regulation". Annual Review of Biochemistry. 65 (1): 337–365. doi:10.1146/annurev.bi.65.070196.002005. ISSN 0066-4154. مؤرشف من الأصل في 12 يوليو 2019. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Dyskeratosis Congenita". NORD (National Organization for Rare Disorders) (باللغة الإنجليزية). مؤرشف من الأصل في 9 يوليو 2019. اطلع عليه بتاريخ 09 أغسطس 2019. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Savage, Sharon A.; Giri, Neelam; Baerlocher, Gabriela M.; Orr, Nick; Lansdorp, Peter M.; Alter, Blanche P. (2008-02). "TINF2, a Component of the Shelterin Telomere Protection Complex, Is Mutated in Dyskeratosis Congenita". The American Journal of Human Genetics. 82 (2): 501–509. doi:10.1016/j.ajhg.2007.10.004. ISSN 0002-9297. مؤرشف من الأصل في 18 ديسمبر 2019. الوسيط
|CitationClass=تم تجاهله (مساعدة); تحقق من التاريخ في:|تاريخ=(مساعدة) - RESERVED, INSERM US14-- ALL RIGHTS. "Orphanet: Dyskeratosis congenita". www.orpha.net (باللغة الإنجليزية). مؤرشف من الأصل في 16 نوفمبر 2017. اطلع عليه بتاريخ 10 أغسطس 2019. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Figure 1: Systems design schematics from: (A) Son et al. (2010); (B) Lau et al. (2010); (C) Fujiwara et al. (2011); and (D) Gjerlufsen et al. (2011)". dx.doi.org. مؤرشف من الأصل في 18 ديسمبر 2019. اطلع عليه بتاريخ 09 يوليو 2019. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Reference, Genetics Home. "Dyskeratosis congenita". Genetics Home Reference (باللغة الإنجليزية). مؤرشف من الأصل في 9 يوليو 2019. اطلع عليه بتاريخ 10 أغسطس 2019. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Dyskeratosis Congenita". NORD (National Organization for Rare Disorders) (باللغة الإنجليزية). مؤرشف من الأصل في 9 يوليو 2019. اطلع عليه بتاريخ 10 أغسطس 2019. الوسيط
|CitationClass=تم تجاهله (مساعدة)
روابط خارجية
- مرض خلل التقرن الخلقي
- GeneReviews / NCBI / NIH / UW entry on Dyskeratosis Congenita
- داء التقرن الخلقي: دراسة بحثية لمتلازمات فشل نخاع العظام الوراثي (IBMFS)
- تتوفر إرشادات DC السريرية، التي كتبها خبراء في هذا المجال، من Dyskeratosis Congenita Outreach.
 بوابة طب
بوابة طب