متلازمة لوجان-فرينس
متلازمة لوجان-فرينس أو متلازمة لوجان أو التخلف العقلي الوراثي السائد المصحوب بأعراض متلازمة مارفان،[1][2][3] هو مرض وراثي سائد على الكروموسوم X الذي يسبب درجات متفاوتة من التخلف العقلي الطفيف إلى المتوسط وبعض الأعراض الشبيهة بأعراض متلازمة مارفان.[4][5] تشمل هذه الأعراض القامة النحيفة الطويلة، والأطراف الطويلة النحيفة.[5] يصاحب متلازمة لوجان-فرينس اضطرابات أخرى في السلوك وبعض الاضطرابات النفسية، بالإضافة إلى بعض التشوهات الدماغية والقلبية.[6][7][8] تتخذ الصورة الوراثية لمتلازمة لوجان-فرينس النمط السائد، ويُعزى هذا إلى طفرة في جين MED12 على الكروموسوم X.[3]
| متلازمة لوجان-فرينس | |
|---|---|
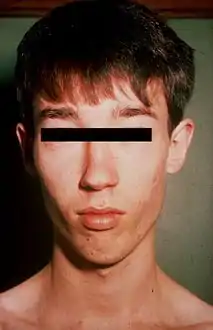 متلازمة لوجان-فرينس في ذكر بالغ صغير، مه وجود بعض الأعراض مثل الوجه الطويل النحيف والذقن المسحوب. متلازمة لوجان-فرينس في ذكر بالغ صغير، مه وجود بعض الأعراض مثل الوجه الطويل النحيف والذقن المسحوب. | |
| معلومات عامة | |
| الاختصاص | طب نفسي |
لا يوجد أي علاج لهذا الخلل الجيني في الوقت الحالي، بينما يظل السبب الريسي لهذا الخلل الجيني مجهولًا حتى وقتنا هذا.[9]
الخصائص
تتراوح درجات التخلف العقلي في متلازمة لوجان-فرينس من التخلف العقلي الطفيف إلى المتوسط، كما تم الإبلاغ عن حدوث حالات شديدة الخطورة من التخلف العقلي كذلك.[10][11] يعتبرعدم تخليق الجسم الثفني من أكثر حالات التشوهات الدماغية شهرةً، وهو ما ينتج عنه خلل في التطور الجنيني للجسم الثفني (وهو جزء من تركيب الدماغ في الثدييات يسمح بانتقال الأعصاب بين نصفي الكرة المخية) مما يؤدي إلى عدم تكوينه.[7][12] يمثل التخلف العقلي من بين الأعراض العصبية المصحابة لعدم تخليق الجسم الثفني، 73% من الأعراض.[12] على الرغم من ذلك لم يتم وضع رابطة بين عدم تخليق الجسم الثفني والتخلف العقلي المصاحب لمتلازمة لوجان-فرينس.[13]
الأعراض النفسية
عادةً ما تظهر الأعراض النفسية والاضطرابات السلوكية في متلازمة لوجان-فرينس، وهو ما يتم أخذه في الحسبان عند التشخيص.[7] من أكثر تلك الأعراض شيوعًا هو طيف التوحد، لذا تعتبر متلازمة لوجان-فرينس هي أحد الأمراض الجينية المصحوبة بالتوحد.[7][14] كان من بين الاضرابات النفسية والسلوكية الأخرى المتواجدة في متلازمة لوجان-فرينس: الذهان[15] والفصام [16] واضطراب نقص الانتباه مع فرط النشاط[13][17] و العدوان [17] و اضطراب التحدي الاعتراضي[13][18] واضطراب الوسواس القهري[13] و الخجل و عجز التعلم [13] و اضطراب الذاكرة قصيرة الأمد [13] و اضطراب القلق الاجتماعي [13] و اضطراب الأكل [13] و سوء التغذية و الأمراض نفسية المنشأ [13] و فقدان الشهية و هوس الحرائق.[7][13][18]
وبينما يتم توقع حدوث مثل تلك الاضطرابات النفسية والسلوكية في حالات الإصابة بمتلازمة لوجان-فرينس، إلا أنه توجد بعض الحالات التي تتمتع بالحفاظ على القوى العقلية والسلوكية والنفسية الطبيعية، مثل حل المشكلات ومستويات الذكاء والفهم الطبيعيين.[19] عادةً ما يصاب أصحاب متلازمة لوجان-فرينس بمرض الفصام. وحيث أن الفصام يعتبر من أحد الأسباب التي تسبب التخلف العقلي، لذا يجب وضع متلازمة لوجان-فرينس في التشخيص التفريقي للفصام مع إجراء الاختبارات الجينية والنفسية اللازمة.[16]
أشباه مارفان
عادةً ما يتم الاعتماد على أعراض متلازمة مارفان لتشخيص حالات لوجان-فرينس وتفرقتها عن الأشكال الأخرى من التخلف العقلي المرتبط بالكروموسوم X.[10]
يصف مصطلح أشباه مارفان بعض الأعراض الطبية الشبيهة بمتلازمة مارفان. تتشارك كلا المتلازمتين بعض الاضطرابات الوراثية الأخرى، أشهرها هو تكون الورم الصماوي المتعدد النوع 2.[20]
تشمل تلك الأعراض الآتي:
- الوجه الطويل النحيف.[5][9]
- القامة الطويلة.[3][9]
- الأطراف والأصابع و أصابع القدم الطويلة النحيفة مع مدى واسع من حركة المفاصل.[3][21][22]
- سلاميات قصيرة لأصابع القدم الكبيرة وطول أصابع القدم الأخرى.
يتأخر تشخيص متلازمة لوجان-فرينس وتفرقتها عن متلازمة مارفان لعدم ظهور العديد من تلك الأعراض حتى سن المراهقة.[2]
الشذوذ القحفي (الوجه والدماغ)
تشمل اضطرابات الوجه والدماغ:
- نقص تنسج الفك العلوي (نقص نمو عظام الفك العلوي)[9]
- الفك السفلي الصغير والذقن المسحوبة.[3][17]
- الحنك ذا التقوس العالي مع ازدحام وعدم ترتيب الأسنان العلوية.[5][7]
- ضخامة الرأس[3][9] (تضخم الجمجمة) وبروز الجبهة وخنف الحديث [5][7] وانخفاض مستوى الأذنين.
- الأنف الطويل وضيق منطقة ما فوق الأنف والنثرة القصيرة (النثرة هي المنخفض الذي يعتلي الشفة العلوية أسفل الأنف)
- نقص التوتر العضلي و الصدر المقعر[9] وتضخم الخصية الطفيف في الذكور ونوبات الصرع.[9][17]
ينتج خنف الحديث من قصور تكوين الخفاق البلعومي، وهو عيب خلقي في تكوين عاصرة الخفاق البلعومي مما يسمح بمرور كمية كبيرة من الهواء إلى جوف الأنف خلال الحديث.[23][24] يمكن أيضًا إرجاع خنف الحديث في متلازمة لوجان-فرينس إلى فشل شراع الحنك واللهاة في الوصول إلى الجانب الخلفي للبلعوم خلال الحديث، وهذه الحالات عادةً ما يصاحبها حالة مرضية تعرف باسم فلح الشفة والحنك.[13][25]
تشوهات القلب والأوعية الدموية
لوحظ العديد من التشوهات القلبية في حالات عديدة مصابة بمتلازمة لوجان-فرينس، أكثرها على الإطلاق كان تمددات شريان الأبهر الصاعد،[26] وتقسم الأبهر الصاعد. يرتبط تمدد الأبهر الصاعد بشكل كبير بتسلخ جدار الأبهر الصاعد، وهو ما ينتج عنه التكيسات الدموية الأبهرية. ونظرًا لتشكيل تلك التشوهات خطرًا على صحة المريض، سرعان ما يتم إجراء تخطيط صدى القلب فور التأكد من تشخيص المتلازمة، جنبًا إلى جنب من إجراء تصوير بالرنين المغناطيسي للدماغ لاحتمالية عدم تخليق الجسم الثفني. كان عيب الحاجز البطيني و عيب الحاجز الأذيني من ضمن التشوهات القلبية الأخرى التي لوحظت في حالات متلازمة لوجان-فرينس.[8][17]
السبب
تم اعتبار حدوث طفرة مغلطة في جين MED12 المتواجد على كروموسوم X سببًا رئيسيًأ لمتلازمة لوجان-فرينس.[3][27] الطفرات المغلطة هي طفرات نقطية حيث يتم إدخال الشيفرة الجينية لحمض أميني من المفترض ألا يتواجد على تتابع الأحماض الأمينية لأحد البروتينات بدلًا من الحمض الأميني المفترض تواجده في هذا التتابع عند نقطة معينة. تحدث الطفرة المغلطة في جين MED12 عند النقطة p.N1007S. وهذا يعني أنه تم استبدال الحمض الأميني أسبارجين، والمفترض تواجده عند الموقع 1007 على جين MED12، بالحمض الأميني سيرين.[27] يتسب هذا الخلل الجيني في تخليق خاطيء للبروتينات المسؤول عنها هذا الجين وبالتالي خطأ في وظيفتها، مما يؤدي إلى تلك الاضطرابات.[3][9]
آلية المرض
يعتبر جين MED12 أحد الجينات المتحكمة في نسخ الحمض النووي الريبوزي الرسول في الثدييات.[28][29]
يعمل هذا الجين كحلقة وصل بين الانزيم وباقي عوامل النسخ المرتبطة بهذا الحمض. يحتوي مركب النسخ هذا على 30 وحدة فرعية، بينما يلزم وجود بعضًا من تلك الوحدات فقط لاتمام تلك العملية في الخلايا والأنسجة المختلفة.[30] في الوقت الحالي، لا تزال الآلية المحددة لحدوث متلازمة لوجان-فرينس والاضطرابات السلوكية والنفسية اللاحقة بها نتيجةً للخلل الجيني في جين MED12 غير واضحة. تظهر أعراض أشباه مارفان مثل الحنك ذو التقوس العالي الخاصة بمتلازمة لوجان-فرينس في متلازمة مارفان وهو أحد أمرض النسيج الضام. ومما يرجح بقوة مشاركة هذ الخلل الجيني في آلية مرض تلك المتلازمة هو تمددات الأبهر الصاعد الذي يوضح خلل في الجين المسؤول عن أحد الأنسجة الضامة.[1][5][8][13]
حصل العلماء على العديد من النتائج المبهرة في دراسة طفرات جين MED12 في الدانيو المخطط، والذي اتخذه العلماء كنموذج حي للفقاريات.[31][32][33] وجد العلماء أن طفرة جين MED12 في ذلك النوع من الأسماك مسؤولة عن طفرة عدم الحركة، وهو ما صاحبها تشوهات عصبية وقلبية، على الرغم من عدم إصابة جميع الأعصاب. عند إعطاء تلك الأسماك حمض نووي ريبوزي رسول الخاص بجين MED12 استعادت تلك الأسماك صحتها. يعتبر جين MED12 أيضًا أحد الجينات اللازمة لتفعيل جين SOX9 اللازم لنمو الغضاريف والعظام والأعصاب.[34][35][36] كان من ضمن التشوهات الناجمة عن هذا الخلل الجيني في تلك الأسماك هو قصور تكوين العرف العصبي، وهو ما أحدث خللًا في الجهاز العصبي الذاتي و الجهاز العصبي المحيطي وتشوهات الخلايا الأم للغضاريف والعظام، وهو ما يماثل عدم تكوين الجسم الثفني وتشوهات الوجه والرأس المرتبطة بالغضاريف والعظام في البشر.[3]
علم الوراثة
 |
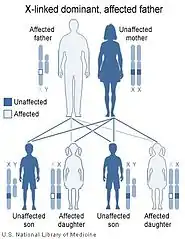 | |
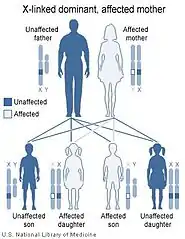
يعتمد انتقال الأمراض الوراثية المرتبطة بالكروموسوم X على حنس الشخص الحامل للمرض إذا كان الأم (كما على اليسار) أو الأب (كما على اليمين). | ||
يتم وراثة متلازمة لوجان-فرينس بصورة سائدة على الكروموسوم X.[9][13][37] وهذا يعني أن الخلل الجيني في جين MED12 متواجد على الكروموسوم X، ووجود نسخة من الجين المختل كافي لوراثة هذا الخلل الجيني من أحد الأبوين. يتمتع الذكور باقترانية زيجوتية للكروموسوم X الذي يمتلكون منه نسخة واحدة. وبالتالي تشيع تلك الحالات بين الذكور أكثر من الإناث.[13][37]
وحيث أن الكروموسوم X هو أحد كروموسومات تحديد الجنس (والآخر هو الكروموسوم Y) ترتبط الصفات الوراثية المنتقلة من خلال الكروموسوم X بجنس الأب الحامل للخل الجيني. يرجع هذا إلى امتلاك الإناث لزوجين من كروموسومات X، بينما يمتلك الذكور نسخةً واحدةً منه. كما يلعب الفرق بين الوراثة السائدة والمتنحية دورًا هامًا في تحديد انتقال الصفات الوراثية من جيل الآباء إلى جيل الأبناء.
في حالات الوراثة السائدة المرتبطة بالكروموسوم X يصبح كلًا من الذكور والإناث معرضان لخطر الإصابة (مع وجود احتمالية إصابة الذكور أعلى من الإناث). يحدث عكس هذا في الوراثة المتنحية المرتبطة بالكروموسوم X، إذ ترتفع نسبة إصابة الإناث عن الذكور لتمتعهم بالافترانية الزيجوتية في تلك الحالة.[13][37]
كما تم الإبلاغ أيضًا عن بعض الحالات التي حدثت بها متلازمة لوجان-فرينس في بعض الذكور دون أن يكون لديهم تاريخ مرضي عائلي بحدوث حالات مشابهة لمتلازمة لوجان-فرينس.[13][15][38]
التشابه بين المتلازمة والأمراض الوراثية الأخرى
وُجدت بعض الحالات المشابهة في التخلف العقلي والأعراض الأخرى لمتلازمة لوجان-فرينس في حالة حدوث حذف في التيلوميرات الفرعية على الموقع الكروموسومي في الكروموسوم 5. صاحب الحذف الجيني في تلك المنطقة في الكروموسوم 5 أعراض مثل التخلف العقلي والاضطرابات النفسية والسلوكية والتوحد وضخامة الرأس وخنف الحديث، بالإضافة إلى متلازمة المواء.[25][39] اقترح فرينس عام 2006 ضرورة اجراء اختبار تفصيلي للكروموسوم 5 باستخدام التهجين الموضعي المتألق للتشخيص التفريقي بينه وبين متلازمة لوجان-فرينس.[9]
كما وجد أن طفرة جين UPF3B على الكروموسوم X ترتبط بالتخلف العقلي.[40] يعتبر جين UPF3B أحد مكونات مركب تحليل الحمض النووي الريبوزي الرسول المسؤول عن مراقبة طفرات ذلك الحمض النووي.ينتج عن الطفرة في ذلك الجين خلل في تلك العملية مما ينتج شرائط قصيرة من الحمض النووي الريبوزي الرسولوما يتبعه من تخليق بروتينات مختلة الوظيفة وما يلحق بها من خلل في النمو البدني والعقلي.[41] تم العثور على أشخاص مصابين بمتلازمة لوجان-فرينس من عائلتين مختلفتين لديهم خلل في جين UPF3B، وهو ما يثبت أن بإمكان تلك المتلازمات الجينية أن تتداخل.[42]
التشخيص
على الرغم من اعتماد تشخيص متلازمة لوجان-فرينس على وجود التخلف العقلي وأعراض أشباه مارفان في المرضى، إلا أنه لا يتم تأكيد التشخيص إلا من خلال إجراء اختبار لتحديد وجود طفرة مغلطة في الموقع p.N1007S في جين MED12.[3][9][10]
التشخيص التفريقي
في التشخيص التفريقي لمتلازمة لوجان-فررينس، تتشابه متلازمة أوبيتز-كافيجيا مع أعراض لوجان-فرينس بالإضافة إلى وجود طفرة مغلطة في جين MED12. من ضمن الأعراض المتشابهة بين المتلازمتين: التخلف العقلي المنتققل عبر الكروموسوم X، فرط النشاط، ضخامة الرأس، عدم تخليق الجسم الثفني، وقلة التوتر العضلي. بينما تتفرد متلازمة أوبيتز-كافيجا بفرط التحدث، فرط القوة في الأنشطة الاجتماعية، رتق الشرج ( انغلاق فتحة الشرج) و فرط تباعد العينين.[43][44]
في حين تتصف متلازمة لوجان-فرينس بوجود طفرة مغلطة في الموقع p.N1007S في جين MED12، تتصف متلازمة أوبيتز-كافيجيا بوجود طفرة مغلطة في الموقع p.R961W في جين MED12.[3][9][13][45]
العلاج
بينما لا يوجد علاج محدد للسبب الوراثي الأساسي لمتلازمة لوجان-فرينس، يمكن أخذ الإجراءات التصحيحية والتدابير الوقائية والتدخلات العلاجية لعلاج مشاكل الوجه والرأس والعظام والاضطرابات النفسية في الاعتبار. كما يجب إجرا التدابير الازمة لمراقبة وعلاج المضاعفات الأكثر خطورة مثل نوبات الصرع والتشوهات القلبية. بالإضافة إلى ذلك، يفضل ينبغي أن يولي الأطباء اهتمام وثيق ورعاية ومتابعة متخصصة لتشخيص ومنع حدوث الاضطرابات النفسية والسلوكية المصاحبة مثل الذهان أو العدوان.[9]
الاحصاءات الوبائية
متلازمة لوجان-فرينس هي مرض وراثي سائد ينتقل عبر الكروموسوم X، وبالتالي يشيع بين الذكور عن الإناث. لم يتم إحصاء نسبة حدوث تلك الحالات بين السكان بشكل دقيق حتى الآن.[9]
التاريخ
سُميت تلك المتلازمة بذلك الاسم وفقًا للطبيبين جون إنريك لوجان وجون بيير فرينس.[21] تم وصف التخلف العقلي والأعراض الشبيهة بمتلازمة مارفان وتشوهات الرأس والدماغ مثل الحنك ذا التقوس العالي بواسطة لوجان وآخرون عام 1984.[17] أجري هذا البحث على أربعة أفراد ذكور في هائلة واحدة تجمعهم قرابة العصب.[3][13][17] كما تم وصف نفس الأعراض في عائلات أخرى، بما في ذلك أخوين، بواسطة فرينس وآخرون في أوائل عام 1987. ومن ثم سميت تلك المتلازمة بمتلازمة لوجان-فرينس.[37]
انظر أيضًا
مراجع
- Lacombe, D.; Bonneau, D.; Verloes, A.; Couet, D.; Koulischer, L.; Battin, J. (1993). "Lujan-Fryns syndrome (X-linked mental retardation with marfanoid habitus): report of three cases and review". Genetic counseling (Geneva, Switzerland). 4 (3): 193–198. ISSN 1015-8146. PMID 8267926. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Fryns, J. P.; Van Den Berghe, H. (1991). "X-linked mental retardation with Marfanoid habitus: a changing phenotype with age?". Genetic counseling (Geneva, Switzerland). 2 (4): 241–244. ISSN 1015-8146. PMID 1799424. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Schwartz, C. E.; Tarpey, P. S.; Lubs, H. A.; Verloes, A.; May, M. M.; Risheg, H.; Friez, M. J.; Futreal, P. A.; Edkins, S.; Teague, J.; Briault, S.; Skinner, C.; Bauer-Carlin, A.; Simensen, R. J.; Joseph, S. M.; Jones, J. R.; Gecz, J.; Stratton, M. R.; Raymond, F. L.; Stevenson, R. E. (July 2007). "The original Lujan syndrome family has a novel missense mutation (p.N1007S) in the MED12 gene". Journal of Medical Genetics. 44 (7): 472–477. doi:10.1136/jmg.2006.048637. ISSN 0022-2593. PMC 2597996. PMID 17369503. الوسيط
|CitationClass=تم تجاهله (مساعدة) - الوراثة المندلية البشرية عبر الإنترنت (OMIM) 154700
- Fryns, J. P.; Buttiens, M.; Opitz, J. M.; Reynolds, J. F. (Oct 1987). "X-linked mental retardation with marfanoid habitus". American Journal of Medical Genetics. 28 (2): 267–274. doi:10.1002/ajmg.1320280202. ISSN 0148-7299. PMID 3322000. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Alonso, P.; Pintos, G.; Almazan, F.; Hernández, L.; Loran, E.; Menchon, J. M.; Vallejo, J. (July 2006). "Eating disorder in a patient with phenotypical features of Lujan-Fryns syndrome". Clinical Dysmorphology. 15 (3): 181–184. doi:10.1097/01.mcd.0000220610.24908.a4. ISSN 0962-8827. PMID 16760741. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Lerma‐Carrillo, I.; Molina, J. D.; Cuevas-Duran, T.; Julve-Correcher, C.; Espejo-Saavedra, J. M.; Andrade-Rosa, C.; Lopez-Muñoz, F. (December 2006). "Psychopathology in the Lujan-Fryns syndrome: report of two patients and review". American Journal of Medical Genetics Part A. 140 (24): 2807–2811. doi:10.1002/ajmg.a.31503. ISSN 1552-4825. PMID 17036352. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Wittine, L. M.; Josephson, K. D.; Williams, M. S. (Oct 1999). "Aortic root dilation in apparent Lujan-Fryns syndrome". American Journal of Medical Genetics. 86 (5): 405–409. doi:10.1002/(SICI)1096-8628(19991029)86:5<405::AID-AJMG2>3.0.CO;2-1. ISSN 0148-7299. PMID 10508979. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Buggenhout, G. V.; Fryns, J. -P. (July 2006). "Lujan-Fryns syndrome (mental retardation, X-linked, marfanoid habitus)". Orphanet Journal of Rare Diseases (Free full text). 1: 26. doi:10.1186/1750-1172-1-26. PMC 1538574. PMID 16831221. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Fryns, J. P.; Buttiens, M.; Van Den Berghe, H. (Jan 1988). "Chromosome X-linked mental retardation and marfanoid syndrome". Journal de Genetique Humaine. 36 (1–2): 123–128. ISSN 0021-7743. PMID 3379374. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Mégarbané A, C. C.; Chammas, C. (1997). "Severe mental retardation with marfanoid habitus in a young Lebanese male. A diagnostic challenge". Genetic Counseling (Geneva, Switzerland). 8 (3): 195–200. ISSN 1015-8146. PMID 9327261. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Jeret, J. S.; Serur, D.; Wisniewski, K. E.; Lubin, R. A. (1987). "Clinicopathological findings associated with agenesis of the corpus callosum". Brain & Development. 9 (3): 255–264. doi:10.1016/s0387-7604(87)80042-6. ISSN 0387-7604. PMID 3310713. الوسيط
|CitationClass=تم تجاهله (مساعدة) - الوراثة المندلية البشرية عبر الإنترنت (OMIM) 309520
- Artigas-Pallarés, J.; Gabau-Vila, E.; Guitart-Feliubadaló, M. (Jan 2005). "Syndromic autism: II. Genetic syndromes associated with autism". Revista de Neurologia. 40 Suppl 1: S151–S162. ISSN 0210-0010. PMID 15736079. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Lalatta, F.; Livini, E.; Selicorni, A.; Briscioli, V.; Vita, A.; Lugo, F.; Zollino, M.; Gurrieri, F.; Neri, G. (Feb 1991). "X-linked mental retardation with marfanoid habitus: first report of four Italian patients". American Journal of Medical Genetics. 38 (2–3): 228–232. doi:10.1002/ajmg.1320380211. ISSN 0148-7299. PMID 2018063. الوسيط
|CitationClass=تم تجاهله (مساعدة) - De Hert, M.; Steemans, D.; Theys, P.; Fryns, J. P.; Peuskens, J. (Apr 1996). "Lujan-Fryns syndrome in the differential diagnosis of schizophrenia". American Journal of Medical Genetics. 67 (2): 212–213. doi:10.1002/(SICI)1096-8628(19960409)67:2<212::AID-AJMG13>3.0.CO;2-M. PMID 8723050. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Lujan, J. E.; Carlin, M. E.; Lubs, H. A.; Opitz, J. M. (Jan 1984). "A form of X-linked mental retardation with marfanoid habitus". American Journal of Medical Genetics. 17 (1): 311–322. doi:10.1002/ajmg.1320170124. ISSN 0148-7299. PMID 6711603. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Williams, M. S. (Dec 2006). "Neuropsychological evaluation in Lujan-Fryns syndrome: commentary and clinical report". American Journal of Medical Genetics Part A. 140 (24): 2812–2815. doi:10.1002/ajmg.a.31501. ISSN 1552-4825. PMID 17103446. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Donders, J.; Toriello, H.; Van Doornik, S. (Jan 2002). "Preserved neurobehavioral abilities in Lujan-Fryns syndrome". American Journal of Medical Genetics. 107 (3): 243–246. doi:10.1002/ajmg.10144. ISSN 0148-7299. PMID 11807907. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Prabhu, M.; Khouzam, R. N.; Insel, J. (Nov 2004). "Multiple endocrine neoplasia type 2 syndrome presenting with bowel obstruction caused by intestinal neuroma: case report". Southern Medical Journal. 97 (11): 1130–1132. doi:10.1097/01.SMJ.0000140873.29381.12. ISSN 0038-4348. PMID 15586612. الوسيط
|CitationClass=تم تجاهله (مساعدة) - synd/3838 على قاموس من سمى هذا؟
- Buntinx, I. M.; Willems, P. J.; Spitaels, S. E.; Van Reempst, P. J.; De Paepe, A. M.; Dumon, J. E. (April 1991). "Neonatal Marfan syndrome with congenital arachnodactyly, flexion contractures, and severe cardiac valve insufficiency". Journal of Medical Genetics. 28 (4): 267–273. doi:10.1136/jmg.28.4.267. ISSN 0022-2593. PMC 1016831. PMID 1856834. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Willging, J. P. (Oct 1999). "Velopharyngeal insufficiency". International Journal of Pediatric Otorhinolaryngology. 49 Suppl 1: S307–S309. doi:10.1016/S0165-5876(99)00182-2. ISSN 0165-5876. PMID 10577827. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Warren, D. W.; Dalston, R. M.; Mayo, R. (Jul 1994). "Hypernasality and velopharyngeal impairment". The Cleft Palate-Craniofacial Journal. 31 (4): 257–262. doi:10.1597/1545-1569(1994)031<0257:HAVI>2.3.CO;2. ISSN 1055-6656. PMID 7918520. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Stathopulu, E.; Ogilvie, C. M.; Flinter, F. A. (June 2003). "Terminal deletion of chromosome 5p in a patient with phenotypical features of Lujan-Fryns syndrome". American Journal of Medical Genetics Part A. 119A (3): 363–366. doi:10.1002/ajmg.a.10268. ISSN 1552-4825. PMID 12784307. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Gambarin, F.; Favalli, V.; Serio, A.; Regazzi, M.; Pasotti, M.; Klersy, C.; Dore, R.; Mannarino, S.; Viganò, M.; Odero, A.; Amato, S.; Tavazzi, L.; Arbustini, E. (April 2009). "Rationale and design of a trial evaluating the effects of losartan vs. Nebivolol vs. The association of both on the progression of aortic root dilation in Marfan syndrome with FBN1 gene mutations". Journal of Cardiovascular Medicine (Hagerstown, Md.). 10 (4): 354–362. doi:10.2459/JCM.0b013e3283232a45. ISSN 1558-2027. PMID 19430350. الوسيط
|CitationClass=تم تجاهله (مساعدة) - الوراثة المندلية البشرية عبر الإنترنت (OMIM) 300188
- Biddick, R.; Young, E. (Sep 2005). "Yeast mediator and its role in transcriptional regulation". Comptes rendus biologies. 328 (9): 773–782. doi:10.1016/j.crvi.2005.03.004. ISSN 1631-0691. PMID 16168358. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Sims, R. J. 3rd; Mandal, S. S.; Reinberg, D. (June 2004). "Recent highlights of RNA-polymerase-II-mediated transcription". Current opinion in cell biology. 16 (3): 263–271. doi:10.1016/j.ceb.2004.04.004. ISSN 0955-0674. PMID 15145350. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Malik, S.; Roeder, R. G. (Jun 2000). "Transcriptional regulation through Mediator-like coactivators in yeast and metazoan cells". Trends in Biochemical Sciences. 25 (6): 277–283. doi:10.1016/S0968-0004(00)01596-6. ISSN 0968-0004. PMID 10838567. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Chakraborty C, H. C.; Hsu, C. H.; Wen, Z. H.; Lin, C. S.; Agoramoorthy, G. (Feb 2009). "Zebrafish: a complete animal model for in vivo drug discovery and development". Current Drug Metabolism. 10 (2): 116–124. doi:10.2174/138920009787522197. ISSN 1389-2002. PMID 19275547. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Kari, G.; Rodeck, U.; Dicker, A. P. (July 2007). "Zebrafish: an emerging model system for human disease and drug discovery". Clinical Pharmacology and Therapeutics. 82 (1): 70–80. doi:10.1038/sj.clpt.6100223. ISSN 0009-9236. PMID 17495877. الوسيط
|CitationClass=تم تجاهله (مساعدة) - McGonnell, I. M.; Fowkes, R. C. (June 2006). "Fishing for gene function--endocrine modelling in the zebrafish". The Journal of Endocrinology. 189 (3): 425–439. doi:10.1677/joe.1.06683. ISSN 0022-0795. PMID 16731775. مؤرشف من الأصل (Free full text) في 15 ديسمبر 2019. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Wang, X.; Yang, N.; Uno, E.; Roeder, R. G.; Guo, S. (November 2006). "A subunit of the mediator complex regulates vertebrate neuronal development". Proceedings of the National Academy of Sciences of the United States of America (Free full text). 103 (46): 17284–17289. Bibcode:2006PNAS..10317284W. doi:10.1073/pnas.0605414103. ISSN 0027-8424. PMC 1859923. PMID 17088561. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Rau, M. J.; Fischer, S.; Neumann, C. J. (Aug 2006). "Zebrafish Trap230/Med12 is required as a coactivator for Sox9-dependent neural crest, cartilage and ear development". Developmental Biology. 296 (1): 83–93. doi:10.1016/j.ydbio.2006.04.437. ISSN 0012-1606. PMID 16712834. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Hong, S. -K.; Haldin, C. E.; Lawson, N. D.; Weinstein, B. M.; Dawid, I. B.; Hukriede, N. A. (December 2005). "The zebrafish kohtalo/trap230 gene is required for the development of the brain, neural crest, and pronephric kidney". Proceedings of the National Academy of Sciences of the United States of America. 102 (51): 18473–18478. Bibcode:2005PNAS..10218473H. doi:10.1073/pnas.0509457102. ISSN 0027-8424. PMC 1311743. PMID 16344459. مؤرشف من الأصل (Free full text) في 15 ديسمبر 2019. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Gurrieri, F.; Neri, G. (Feb 1991). "A girl with the Lujan-Fryns syndrome". American Journal of Medical Genetics. 38 (2–3): 290–291. doi:10.1002/ajmg.1320380225. ISSN 0148-7299. PMID 2018074. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Fryns, J. P. (Feb 1991). "X-linked mental retardation with marfanoid habitus". American Journal of Medical Genetics. 38 (2–3): 233–233. doi:10.1002/ajmg.1320380212. ISSN 0148-7299. PMID 2018064. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Fang, J. S.; Lee, K. F.; Huang, C. T.; Syu, C. L.; Yang, K. J.; Wang, L. H.; Liao, D. L.; Chen, C. H. (Jun 2008). "Cytogenetic and molecular characterization of a three-generation family with chromosome 5p terminal deletion". Clinical Genetics. 73 (6): 585–590. doi:10.1111/j.1399-0004.2008.00995.x. ISSN 0009-9163. PMID 18400035. الوسيط
|CitationClass=تم تجاهله (مساعدة) - الوراثة المندلية البشرية عبر الإنترنت (OMIM) 300298
- Chang, Y. F.; Imam, J. S.; Wilkinson, M. F. (2007). "The nonsense-mediated decay RNA surveillance pathway". Annual Review of Biochemistry. 76: 51–74. doi:10.1146/annurev.biochem.76.050106.093909. ISSN 0066-4154. PMID 17352659. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Tarpey, P. S.; Raymond, F. L.; Nguyen, L. S.; Rodriguez, J.; Hackett, A.; Vandeleur, L.; Smith, R.; Shoubridge, C.; Edkins, S.; Stevens, C.; O'Meara, S.; Tofts, C.; Barthorpe, S.; Buck, G.; Cole, J.; Halliday, K.; Hills, K.; Jones, D.; Mironenko, T.; Perry, J.; Varian, J.; West, S.; Widaa, S.; Teague, J.; Dicks, E.; Butler, A.; Menzies, A.; Richardson, D.; Jenkinson, A.; Shepherd, R. (September 2007). "Mutations in UPF3B, a member of the nonsense-mediated mRNA decay complex, cause syndromic and nonsyndromic mental retardation". Nature Genetics (Free full text). 39 (9): 1127–1133. doi:10.1038/ng2100. ISSN 1061-4036. PMC 2872770. PMID 17704778. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Graham, J. M.; Superneau, D.; Rogers, R. C.; Corning, K.; Schwartz, C. E.; Dykens, E. M. (1999). "Clinical and behavioral characteristics in FG syndrome". American Journal of Medical Genetics. 85 (5): 470–475. doi:10.1002/(SICI)1096-8628(19990827)85:5<470::AID-AJMG7>3.0.CO;2-S. PMID 10405444. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Jr, G.; Visootsak, M.; Dykens, J.; Huddleston, E.; Clark, L.; Jones, R. D.; Moeschler, K. L.; Opitz, J. B.; Morford, J. M.; Simensen, R.; Rogers, R. C.; Schwartz, C. E.; Friez, M. J.; Stevenson, R. E. (December 2008). "Behavior of 10 patients with FG Syndrome (Opitz-Kaveggia Syndrome) and the p.R961W Mutation in the MED12 Gene". American Journal of Medical Genetics Part A. 146A (23): 3011–3017. doi:10.1002/ajmg.a.32553. ISSN 1552-4825. PMC 3092600. PMID 18973276. الوسيط
|CitationClass=تم تجاهله (مساعدة) - الوراثة المندلية البشرية عبر الإنترنت (OMIM) 305450
مصادر خارجية
- GeneReview/NIH/UW entry on MED12-Related Disorders
- Van Buggenhout, G. J. C. M.; Trommelen, J. C. M.; Brunner, H. G.; Hamel, B. C. J.; Fryns, J. P. (Jan 2001). "The clinical phenotype in institutionalised adult males with X-linked mental retardation (XLMR)". Annales de Génétique. 44 (1): 47–55. doi:10.1016/S0003-3995(01)01038-3. ISSN 0003-3995. PMID 11334618.
 بوابة طب
بوابة طب