كثرة الكريات الحمر الكروية الوراثية
كثرة الكريات الحمراء الكروية الوراثي (بالإنجليزية: Hereditary spherocytosis) (اختصارًا HS) هو مرض وراثي، يحدث هذا الاضطراب بسبب وجود خلل في البروتينات التي تشكل غشاء خلايا الدم الحمراء التي تصبح كروية الشكل وهشة، والذي ينجم عن تدمير وتلف كريات الدم الحمراء في الطحال. مما يؤدي إلى فقر الدم الانحلالي المزمن.[1] تم اكتشافه لأول مرة في عام 1871. وهو السبب الأكثر شيوعًا لانحلال الدم الوراثي عند سكان القوقاز في أوروبا وأمريكا الشمالية، مع حدوث 1 لكل 5000 ولادة. وتشمل الأعراض فقر الدم واليرقان، تضخم الطحال، والتعب.[2] علاوة على ذلك، تتراكم بقايا خلايا الدم المكسورة - البيليروبين غير المقرون أو غير المباشر - في المرارة، ويمكن أن تتسبب في تطور حصوات المرارة المصطبغة. لدى المرضى المزمنة، يمكن أن تسبب العدوى أو غيرها من الأمراض زيادة في تدمير خلايا الدم الحمراء، مما يؤدي إلى ظهور أعراض حادة، أزمة الانحلالية.
| كثرة الكريات الحمر الكروية الوراثية | |
|---|---|
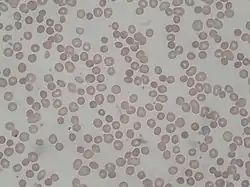 تشويه الدم المحيطي من المريض المصاب بالكثرة الكريات الحمر تشويه الدم المحيطي من المريض المصاب بالكثرة الكريات الحمر | |
| معلومات عامة | |
| الاختصاص | علم الدم |
| من أنواع | فقر الدم الانحلالي الخلقي ، وكثرة الكريات الحمر الكروية |
كان العلاج الأساسي للمرضى الذين يعانون من أعراض HS هو استئصال الطحال الكلي، مما يلغي عملية الانحلال، ويسمح بمستويات الهيموغلوبين الطبيعية، والبيليروبين. يمكن أن تهدد الحالات الحادة بالتسبب في نقص الأكسجة من خلال فقر الدم والتهاب الكبد الحاد من خلال مستويات الدم المرتفعة من البيليروبين، وخاصة عند الأطفال حديثي الولادة. يمكن اكتشاف معظم الحالات بعد الولادة بفترة وجيزة. ينبغي على البالغين المصابين بهذا المرض اختبار أطفالهم، على الرغم من أن وجود هذا المرض عند الأطفال عادةً ما يتم ملاحظته بعد الولادة بفترة قصيرة. في بعض الأحيان، لن يمر المرض دون أن يلاحظه أحد حتى يبلغ عمر الطفل حوالي 4 أو 5 سنوات. قد يكون الشخص أيضًا حاملًا للمرض ولا تظهر عليه أي علامات أو أعراض للمرض. قد تشمل الأعراض الأخرى ألم بطني قد يؤدي إلى إزالة الطحال أو المرارة.
مضاعفات
كثرة الكريات الحمراء الوراثي هو مرض حميدة عموما قد يتواقع ظهوره في متوسط العمر تقريبا. اليرقان يتقلب ويزداد سوءًا في ظروف معينة، خاصة أثناء الحمل والإرهاق البدني والتعرض للبرد والعواصف. في كل الحالات، قد لا يظهر المرض أي علامات ولا يتم اكتشافه إلا أثناء إجراء تقييم منهجي. ومع ذلك يتعرض المريض لبعض المضاعفات:
- قد تحدث هجمات فرط التحلل الشديد بشكل تلقائي أو في ظل الظروف المذكورة أعلاه. الأعراض هي أعراض أي أزمة انحلالية (قشعريرة، ارتفاع في درجة الحرارة، ضيق التنفس، خفقان، ألم وتورم في الطحال والكبد، انخفاض حاد في خلايا الدم الحمراء التي يمكن أن تنخفض إلى أقل من مليون لكل ملليمتر مكعب). يمكن أن تصبح إحدى الأزمات قاتلة، لكن التجديد أمر معتاد.
- أزمة مرنة مع انخفاض كبير في مستوى الهيموغلوبين و (عدد الخلايا الشبكية)، عادة بسبب توقف النضج وغالبًا ما يرتبط بالتغيرات الضخمة. قد يكون عجل من العدوى، مثل الأنفلونزا، وخاصة مع فيروسات برفوف B19.[3][4][5]
- نقص حمض الفوليك الناجم عن زيادة متطلبات نخاع العظام.
- تحدث حصوات المرارة المصطبغة عند حوالي نصف المرضى غير المعالجين. زيادة انحلال خلايا الدم الحمراء يؤدي إلى زيادة مستويات البيليروبين، لأن البيليروبين هو نتاج انهيار الهيم. يجب أن تفرز مستويات عالية من البيليروبين في الصفراء عن طريق الكبد، مما قد يتسبب في تشكيل حصاة مصطبغة، والتي تتكون من البيليروبين الكالسيوم. نظرًا لأن هذه الحجارة تحتوي على مستويات عالية من كربونات الكالسيوم والفوسفات، فهي عبارة عن أقطاب الأشعة وتكون مرئية على الأشعة السينية.
- قرحة الساق.
- فشل القلب يمكن أن يحدث نتيجة لفقر الدم المزمن.
علاج
على الرغم من أن الأبحاث لا تزال جارية، في هذه المرحلة لا يوجد علاج للخلل الوراثي الذي يسبب كثرة الكريات البيضاء وراثيًا.[6] تركز الإدارة الحالية على التدخلات التي تحد من شدة المرض. خيارات العلاج تشمل:
- استئصال الطحال: (الاستئصال الجراحي للطحال) كما هو الحال في كثرة الكريات البيضاء غير الوراثية، تشير الأعراض الحادة لفقر الدم وفرط بيليروبين الدم إلى العلاج بنقل الدم أو التبادل والأعراض المزمنة لفقر الدم والطحال الموسع تشير إلى المكملات الغذائية لحمض الفوليك واستئصال الطحال، يشار إلى استئصال الطحال للحالات المعتدلة إلى الشديدة، ولكن ليس الحالات الخفيفة.[7] لتقليل خطر التسمم، يحتاج مرضى كثرة الكريات بعد استئصال الطحال إلى التمنيع ضد فيروس الأنفلونزا، والبكتيريا المغلفة مثل العقدية الرئوية والمكورات السحائية، والعلاج بالمضادات الحيوية الوقائية. ومع ذلك، لا يزال استخدام المضادات الحيوية الوقائية، مثل البنسلين، مثيراً للجدل.[6]
- استئصال الطحال الجزئي: نظرًا لأن الطحال مهم للحماية من الكائنات المغلفة، فإن التسمم الناجم عن الكائنات المغلفة يعد أحد المضاعفات المحتملة لاستئصال الطحال.[2] يمكن النظر في خيار استئصال الطحال الجزئي لصالح الحفاظ على وظيفة المناعة. البحث عن النتائج محدود حاليًا،[2] ولكنه مواتٍ.[8]
- الاستئصال الجراحي للمرارة قد يكون ضروريًا.[6]
علم الأوبئة
كثرة الخلايا الحمراء الكروية الوراثية هي أكثر اضطرابات الغشاء الخلوي شيوعًا وتؤثر على شخص واحد من كل 2000 شخص من أصل أوروبي شمالي.[9]
البحوث
يوجد علاج جيني تجريبي لعلاج كثرة الكريات الحمراء الوراثية عند الفئران المعملية؛ ومع ذلك، لم يتم بعد تجربة هذا العلاج على البشر بسبب جميع المخاطر التي ينطوي عليها العلاج الجيني البشري.
انظر أيضًا
مراجع
- Cotran, Ramzi S.; Kumar, Vinay; Fausto, Nelson; Nelso Fausto; Robbins, Stanley L.; Abbas, Abul K. (2005). Robbins and Cotran pathologic basis of disease. St. Louis, Mo: Elsevier Saunders. صفحة 625. ISBN 0-7216-0187-1. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Bolton-Maggs, P. H. B.; Stevens, R. F.; Dodd, N. J.; Lamont, G.; Tittensor, P.; King, M. -J.; General Haematology Task Force of the British Committee for Standards in Haematology (2004). "Guidelines for the diagnosis and management of hereditary spherocytosis". British Journal of Haematology. 126 (4): 455–474. doi:10.1111/j.1365-2141.2004.05052.x. PMID 15287938. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Fjaerli, H. O.; Vogt, H.; Bruu, A. L. (1991). "Human parvovirus B19 as the cause of aplastic crisis in hereditary spherocytosis". Tidsskrift for den Norske Laegeforening : Tidsskrift for Praktisk Medicin, NY Raekke. 111 (22): 2735–2737. PMID 1658972. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Beland, S. S.; Daniel, G. K.; Menard, J. C.; Miller, N. M. (1997). "Aplastic crisis associated with parvovirus B19 in an adult with hereditary spherocytosis". The Journal of the Arkansas Medical Society. 94 (4): 163–164. PMID 9308316. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Servey, J. T.; Reamy, B. V.; Hodge, J. (2007). "Clinical presentations of parvovirus B19 infection". American Family Physician. 75 (3): 373–376. PMID 17304869. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Anthony S. Fauci; Eugene Braunwald; Dennis L. Kasper; Stephen L. Hauser; Dan L. Longo; J. Larry Jameson; Joseph Loscalzo (2008). Harrison's principles of internal medicine (الطبعة 17th). New York: McGraw-Hill Medical. صفحات Chapter 106. ISBN 978-0071466332. مؤرشف من الأصل في 7 مايو 2020. اطلع عليه بتاريخ أكتوبر 2020. الوسيط
|CitationClass=تم تجاهله (مساعدة); تحقق من التاريخ في:|تاريخ الوصول=(مساعدة) - Bolton-Maggs PH, Stevens RF, Dodd NJ, Lamont G, Tittensor P, King MJ (أغسطس 2004). "Guidelines for the diagnosis and management of hereditary spherocytosis". Br. J. Haematol. 126 (4): 455–74. doi:10.1111/j.1365-2141.2004.05052.x. PMID 15287938. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Buesing, K. L.; Tracy, E. T.; Kiernan, C.; Pastor, A. C.; Cassidy, L. D.; Scott, J. P.; Ware, R. E.; Davidoff, A. M.; Rescorla, F. J.; Langer, J. C.; Rice, H. E.; Oldham, K. T. (2011). "Partial splenectomy for hereditary spherocytosis: A multi-institutional review". Journal of Pediatric Surgery. 46 (1): 178–183. doi:10.1016/j.jpedsurg.2010.09.090. PMID 21238662. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Hereditary spherocytosis" en (باللغة الإنجليزية). مؤرشف من الأصل في 28 يوليو 2018. اطلع عليه بتاريخ 23 ديسمبر 2019. الوسيط
|CitationClass=تم تجاهله (مساعدة); Invalid|script-title=: missing prefix (مساعدة)
روابط خارجية
 بوابة طب
بوابة طب
 صور وملفات صوتية من كومنز
صور وملفات صوتية من كومنز