داء ليبوفوسيني سيرويدي عصبي
الداء الليبوفوسيني السيرويدي العصبي (بالإنجليزية: Neuronal ceroid lipofuscinosis، ومختصره CLN) هو أو العته الكمني هو اسم عام لعائلة تتكون على الأقل من ثمانية اضطرابات تنكّسية عصبية خازنة للجسيمات منفصلة جينيا، تنتج عن تراكم فائض لصباغ شحمي يدعى ليبوفوسين في أنسجة الجسم.[1]
| الداء الليبوفوسيني السيرويدي العصبي | |
|---|---|
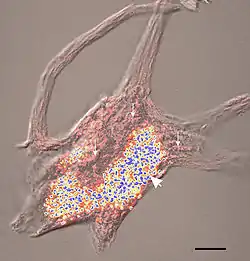 التراكيب الصفراء والزرقاء هي حبيبات ليبوفوسين التراكيب الصفراء والزرقاء هي حبيبات ليبوفوسين | |
| معلومات عامة | |
| الاختصاص | علم الغدد الصم |
| من أنواع | مرض الجهاز العصبي الوراثي التنكسي |
تتكوّن هذه الأصبغة الشحمية من دهون وبروتينات، تأخذ لونًا أصفر مُخضَر عند رؤيتها تحت مجهر يستخدم الأشعة فوق البنفسجيّة. تتراكم المواد الدهنية هذه في الخلايا العصبية والعديد من الأعضاء، بما في ذلك الكبد والطحال وعضلة القلب والكلى.
العلامات والأعراض
تتميّز مجموعة اضطرابات التخزين العصبي الليزوزيني الذي يطلق عليه اسم الداء الليبوفوسيني السيرويدي العصبي، بالفقدان التدريجي والدائم للقدرات الحركيّة والنفسيّة، مع تراكم شديد لليبوفوسين بين الخلايا.[2][3] يحدث المرض بمعدّل أعلى بقليل في أوروبا الشماليّة والولايات المتّحدة الأمريكيّة، بمعدّل حدوث 1 من كل 10000 شخص.[4] هناك أربعة تشخيصات كلاسيكية حظيت بأكبر قدر من الاهتمام من الباحثين والعاملين في المجال الطبي، تختلف عن بعضها البعض حسب عمر بدء الأعراض، والمدة، والمظاهر السريريّة عند البداية المبكرة للمرض مثل العمى أو الاختلاجات، والأشكال التي يأخذها تراكم الليبوفوسين.[2]
في الداء الليبوفوسيني السيرويدي الطفلي، يكون الطفل طبيعيًا عند الولادة، ولا يوجد أي عرض مُلفِت للنظر. فقدان البصر الباكر، الذي يؤدي إلى عمى شبكيّ بعمر السنتين، هو الموشّر الأوّل للمرض. بعمر الثلاث سنوات، يصل الطفل إلى الحالة الإنباتيّة، وبعمر الأربع سنوات، يُثبت مخطّط كهربية الدماغ الكهرساوي موت الدماغ.
بينما يتظاهر الشكل الطفلي المتأخّر من الداء الليبوفوسيني السيرويدي العصبي، بعمر الاثنين إلى أربع سنوات، على شكل اختلاجات وتدهور في القدرة البصريّة، العمر الأقصى للطفل المصاب بالشكل المتأخر هو 10-12 عام.[5][6][7][8]
يتظاهر الداء الليبوفوسيني السيرويدي العصبي اليُفعي، لأول مرّة بعمر 4-10 سنوات، على شكل فقدان مهم بالقدرة البصريّة، واختلاجات، وتدهور الحالة النفسيّة، والموت بعمر 20-30.[9]
يتظاهر الداء الليبوفوسيني السيرويدي العصبي عند البالغين (داء كوف)، لأول مرّة بعمر 30 عام، بأعراض أكثر اعتدالاً وأخفّ من الحالات السابقة، ولكنّه مفهوم بشكل أقل منها، يحدث الموت بعد عشر سنوات من بدء الأعراض.[1]
تبيَّن أنّ جميع الطفرات المرتبطة بالمرض، لها علاقة بعمليات الاستقلاب في منطقة المشابك العصبية، وخاصةً إعادة استخدام الحويصلات البروتينية.
الوراثة
الداء الليبوفوسيني السيرويدي عند الأطفال هو اضطراب وراثي جسمي متنحٍ، أي يحدث عندما يرث الطفل نسختين من الجين المصاب، من كل من الوالدين، عندما يحمل كل من الوالدين جيناً واحداً معيباً، تكون فرصة إصابة الأطفال واحد إلى أربعة، بينما يبلغ احتمال وراثة الطفل لجين واحد معيب واحد إلى اثنان، يُعرَّف الأفراد الحاملون لجين واحد معيب بأنهم حاملون للمرض، أي أنّهم غير مُصابين، ولكنّهم يستطيعون نقل المرض إلى الأبناء. تحدث الطفرات الأكثر تمييزاً للمرض في «جين CLN3». يقع هذا الجين على الذراع القصير للصبغي 16. الوظيفة الطبيعية للجين غير معروفة حالياً ولكنها تُرمّز بروتين عابر للغشاء.
يُورَّث الداء الليبوفوسيني السيرويدي العصبي، إما كاضطراب جسمي متنحٍ، وقلّما كاضطراب جسمي سائد، في الوراثة الجسمية السائدة، يُصاب جميع الأشخاص الذين يرثون نسخة واحدة من الجين المعيب. وبالتالي، لا يوجد أشخاص حاملون للمرض غير مصابين.
يعتمد الكثيرون على تسميّة مجموعة أمراض الداء الليبوفوسيني السيرويدي العصبي ب «داء باتن»
التشخيص
نظرًا لأن فقدان البصر غالباً ما يكون علامة مبكرة في الداء الليبوفوسيني السيرويدي العصبي، يُشتبه بالمرض أولاً عند فحص العين، إذ يلاحظ طبيب أمراض العين فقداناً في الخلايا البصرية الذي تحدث في أشكال الطفولة الثلاثة، ولكنّ فقدان الخلايا يحدث في أمراض أخرى، وبالتأكيد لا يكفي للتشخيص. عندما يشتبه طبيب أمراض العين بالمرض ينبغي دوماً إحالة الطفل إلى طبيب مختص بالأمراض لعصبية، إذ يقوم الأخير بأخذ قصّة سريريّة مفصّلة، وفحوص مخبريّة.
تتضمّن الاختبارات التشخيصية المستخدمة في الداء الليبوفوسيني السيرويدي العصبي ما يلي:
- أخذ عينات من الجلد أو الأنسجة: يمكن للطبيب فحص قطعة صغيرة من الأنسجة تحت المجهر الإلكتروني. يساعد التكبير القوي للمجهر الطبيب على اكتشاف رواسب الليبوفوسين النموذجية. توجد هذه الرواسب في العديد من الأنسجة المختلفة، بما في ذلك الجلد والعضلات والملتحمة والمستقيم وغيرها. يمكن أيضًا استخدام الدم. تأخذ هذه الرواسب أشكالًا مميزة للمرض.
- مخطط كهربية الدماغ: يستخدم مخطّط كهربيّة الدماغ مسارٍ خاصة، تُوضَع على فروة الرأس لتسجيل التيارات الكهربائية داخل المخ. يساعد مخطط كهربيّة الدماغ الأطباء على تحديد أنماط النشاط الكهربائي في الدماغ، التي قد تُشير إلى إصابة المريض باختلاج مثلاً.
- دراسات كهربية العينين: يمكن لهذه الاختبارات، التي تشمل الاستجابة البصريّة المُستثارة، ومخطّط كهربيّة الشبكية، أن تكشف اضطرابات العين المختلفة المرافقة للمرض.
- مسح الدماغ: يمكن أن يساعد التصوير الأطباء في البحث عن التغييرات في مظهر الدماغ. تقنية التصوير الأكثر استخدامًا هي التصوير المقطعي، الذي يستخدم الأشعة السينية وجهاز الكمبيوتر لإنشاء صورة متطوّرة لأنسجة الدماغ وتراكيبه. يكشف التصوير المقطعي مناطق الدماغ الآخذة بالتلاشي. تقنية التصوير الثانية الشائعة بشكل متزايد هي التصوير بالرنين المغناطيسي. يستخدم التصوير بالرنين المغناطيسي مجموعة من الحقول المغناطيسية وموجات الراديو، بدلاً من الإشعاع، لإنشاء صورة للمخ.
- مُقايسة الإنزيم: من التطورات الحديثة في تشخيص الداء الليبوفوسيني السيرويدي العصبي هو استخدام مقايسة الإنزيم، للبحث عن إنزيمات ليزوزومية مفقودة في الطفولة والطفولة المتأخرة فقط. تعتبر المقايسة الإنزيمية اختبار تشخيصي سريع وسهل.
العلاج
لا يوجد حالياً علاج مقبول على نطاق واسع يمكنه علاج أو إبطاء أو إيقاف الأعراض لدى الغالبية العظمى من المرضى الذين يعانون من الداء الليبوفوسيني السيرويدي العصبي. ومع ذلك، يمكن السيطرة على الاختلاجات أو تخفيضها باستخدام الأدوية المضادة للصرع، وقد تساعد العلاجات الجسدية والكلامية والمهنية المرضى المصابين، للحفاظ على وظائفهم لأطول فترة ممكنة، وهناك العديد من العلاجات التجريبية قيد الدراسة.
- سيستاغون: تبيّن أنّ هذا العقّار يبطّئ عمليّة التنكّس العصبي عند المرضى المصابين بالداء الليبوفوسيني السيرويدي العصبي الطفلي.
- المعالجة الجينيّة: بدأت تجارب المعالجة الجينيّة لهذا المرض، في يونيو من عام 2004، كمحاولة لمعالجة الأشكال المتأخرة من الداء الليبوفوسيني السيرويدي العصبي.
- فلابيريتين.
- المعالجة بالخلايا الجذعية.
- كابحات المناعة.
- لعلاج ببدائل الإنزيم.
الأنواع
التصنيف القديم للداء الليبوفوسيني السيرويدي العصبي يقسمه إلى أربعة آنواع (CLN1 و CLN2 و CLN3 و CLN4) اعتمادا على العمر الذي تبدأ فيه الأعراض، في المقابل يعتمد التقسيم الجديد على الجين المسيبب للمرض.[10][11]
النوع الرابع (CLN4) لم يحدد فيه نوع جين معين بخلاف الأنواع البقية (CLN1 و CLN2 و CLN3).
ينقسم الداء الليبوفوسيني السيرويدي العصبي بحسب التصنيف القديم إلى أربعة أنواع، اعتماداً على العمر الذي تبدأ فيه الأعراض، في المقابل يعتمد التقسيم الجديد على الجين المسبّب للمرض.[12][11]
لم يُحدَّد الجين المُصاب في النوع الرابع بخلاف الأنواع البقية.
| النوع | الوصف | الوراثة المندلية البشرية عبر الإنترنت | الجين |
| النوع الأول | الداء الليبوفوسيني السيرويدي العصبي الطفلي (INCL) أو داء سانتافوري-هالتيا أو داء سانتافوري: يبدأ بعمر ما بين 6 أشهر وسنتين ويتفاقم بسرعة. | 256730 | بلميتول-بروتين ثيوسترس 1 |
| النوع الثاني | الداء الليبوفوسيني السيرويدي العصبي الطفلي الآجل (LINCL) أو داء يانسكي–بيلشوفسكي، تبدأ الأعراض بين عمر السنتين و4 سنوات. يتقدم هذا النوع بسرعة وينتهي بالوفاة بين عمري 8 سنوات 12 سنة. | 204500 | TPP1 |
| النوع الثالث | الداء الليبوفوسيني السيرويدي العصبي اليفعي (JNCL) أو داء باتن، يبدأ بين عمري 5 سنوات و8 سنوات. يتقدم هذال النوع بسرعة أقل وينتهي بالوفاة في فترة العشرينات من العمر، لكن هنالك حالات عاشت حتى الثلاثينات من العمر. | 204200 | CLN3 |
| النوع الرابع | الداء الليبوفوسيني السيرويدي العصبي للبالغين (ANCL) أو داء كوفس، يبدأ عادة قبل سنة 40 من العمر، يتسبب بأعراض أخف ويتفاقم ببطء ولا يتسبب بالعمى. عادة ما يكون سن الوفاة مختلفا للمصابين بهذا المرض، ويبدو أنه لا يتسبب بتقليل العمر المتوقع للوفاة. | 204300 (AR), 162350 (AD) | CLN6[13] DNAJC5 |
| النوع الخامس | الداء الليبوفوسيني السيرويدي العصبي الطفلي الفنلندي الآجل (vLINCL) – شخص في فنلندا. | 256731 | CLN5 |
| النوع السادس | الداء الليبوفوسيني السيرويدي العصبي الطفلي الآجل المختلف (vLINCL) – شخص في كوستاريكا وأمريكا الجنوبية والبرتغال والمملكة المتحدة ودول أخرى. | 601780 | CLN6 |
| النوع السابع | CLN7 | 610951 | MFSD8 |
| النوع الثامن | متلازمة الصرع الشمالية أو الصرع المتقدم مع التخلف العقلي (EPMR) - CLN8 | 610003 | CLN8 |
| النوع الثامن | النوع التركي الطفلي (vLINCL) – شخص في تركيا. | 600143 | CLN8 |
| النوع التاسع | شخص في ألمانيا وصربيا. | 609055 | غير معروف، لكن يحتمل منظم لثنائي هيدرو-سيراميد سينثاز[14] |
| النوع العاشر | CLN10 (خلقي، نقص كاثبسين د) | 116840 | CTSD |
مراجع
- Mole, Sara E.; Williams, Ruth E. (1 January 1993). "Neuronal Ceroid-Lipofuscinoses". GeneReviews. مؤرشف من الأصل في 13 يونيو 2019. اطلع عليه بتاريخ 11 ديسمبر 2016. الوسيط
|CitationClass=تم تجاهله (مساعدة)update 2013 - Pardo, C.; et al. (1994). "Accumulation of the adenosine triphosphate synthase subunit c in the mnd mutant mouse". Am J Pathol. 144 (4): 829–835. PMC 1887237. PMID 8160780.
There are more than eight variants of NCL, found in 1 in 12,500 people worldwide.
الوسيط|CitationClass=تم تجاهله (مساعدة) - Narayan SB, Pastor JV, Mitchison HM, Bennett MJ (Aug 2004). "CLN3L, a novel protein related to the Batten disease protein, is overexpressed in Cln3-/- mice and in Batten disease". Brain. 127 (Pt 8): 1748–54. doi:10.1093/brain/awh195. PMID 15240430. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Vesa J, Chin MH, Oelgeschläger K, et al. (Jul 2002). "Neuronal ceroid lipofuscinoses are connected at molecular level: interaction of CLN5 protein with CLN2 and CLN3". Mol Biol Cell. 13 (7): 2410–20. doi:10.1091/mbc.E02-01-0031. PMC 117323. PMID 12134079. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Hellstein, E.; et al. (1996). "Human palmitoyl protein thioesterase". Eur Mol Bio Org J. 15 (19): 5240–5. doi:10.1002/j.1460-2075.1996.tb00909.x. PMC 452268. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Kim, S.; et al. (2006). "PPT1 deficiency leads to the activation of caspase-9 and contributes to rapid neurodegeneration in INCL". Hum Mol Genet. 15 (10): 1586–90. doi:10.1093/hmg/ddl078. PMID 16571600.
Late Infantile NCL (LINCL or Jansky-Bielschowsky), on the other hand, initially presents as generalized tonic-clonic or myoclonic seizures beginning at around 2–3 years of age; following this is depressed cognitive development including slow learning, speech delays, and eventual dementia leading to death, usually between 14 and 36 years of age.
الوسيط|CitationClass=تم تجاهله (مساعدة) - Ju, W.; et al. (2002). "Identification of CLN2 mutations shows Canadian specific NCL2 alleles". Journal of Medical Genetics. 39 (11): 822–825. doi:10.1136/jmg.39.11.822. PMC 1735024. PMID 12414822. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Isosomppi, J.; et al. (2002). "Lysosomal localization of the neuronal ceroid lipofuscinosis CLN5 protein". Hum Mol Genet. 11 (8): 885–91. doi:10.1093/hmg/11.8.885. PMID 11971870. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Persaud-Sawin, D.; et al. (2002). "Motifs within the CLN3 protein". Hum Mol Genet. 11 (18): 2129–2142. doi:10.1093/hmg/11.18.2129. PMID 12189165. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Mole SE, Williams RE, Goebel HH (September 2005). "Correlations between genotype, ultrastructural morphology and clinical phenotype in the neuronal ceroid lipofuscinoses". Neurogenetics. 6 (3): 107–26. doi:10.1007/s10048-005-0218-3. PMID 15965709. الوسيط
|CitationClass=تم تجاهله (مساعدة) - الوراثة المندلية البشرية عبر الإنترنت (OMIM) 256730
- Mole SE, Williams RE, Goebel HH (September 2005). "Correlations between genotype, ultrastructural morphology and clinical phenotype in the neuronal ceroid lipofuscinoses". Neurogenetics. 6 (3): 107–26. doi:10.1007/s10048-005-0218-3. PMID 15965709. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Arsov, T; et al. (13 May 2011). "Kufs Disease, the Major Adult Form of Neuronal Ceroid Lipofuscinosis, Caused by Mutations in CLN6". American Journal of Human Genetics. 88 (5): 566–73. doi:10.1016/j.ajhg.2011.04.004. PMC 3146726. PMID 21549341. الوسيط
|CitationClass=تم تجاهله (مساعدة);|access-date=بحاجة لـ|url=(مساعدة) - Schulz A, Mousallem T, Venkataramani M, et al. (February 2006). "The CLN9 protein, a regulator of dihydroceramide synthase". J. Biol. Chem. 281 (5): 2784–94. doi:10.1074/jbc.M509483200. PMID 16303764. مؤرشف من الأصل في 18 ديسمبر 2019. الوسيط
|CitationClass=تم تجاهله (مساعدة)
 بوابة طب
بوابة طب بوابة علوم عصبية
بوابة علوم عصبية