داء باتن
مرض باتن أو داء باتن (بالإنجليزية: Batten disease) هو مرض وراثي نادر، يتسبب في اضطرابات على مستوى الجهاز العصبي وتدهور متواصل لأجهزة جسم المريض، تؤدي به في النهاية إلى العمى وفقدان النطق والشلل، ومن ثم الموت المبكر. تظهر أولى أعراض المرض عادة خلال مرحلة الطفولة، في سن يتراوح بين 5 و 10 سنوات.[1] يمثل مرض باتن الشكل الأكثر شيوعا لمجموعة من الاضطرابات التنكسية العصبية، التي يطلق عليها اسم "الليفوسينات السيرويدية العصبية" (NCLs).
| داء باتن Batten disease | |
|---|---|
| معلومات عامة | |
| الاختصاص | علم الغدد الصم |
| من أنواع | داء ليبوفوسيني سيرويدي عصبي |
تاريخ
وصف مرض باتن لأول مرة في سنة 1903 على يد طبيب الأطفال البريطاني فريدريك يوستاس باتن.[2] بحلول نوفمبر لسنة 1905 أبلغ الطبيب الألماني فالتر سبيميلير عن ثلاثة حالات لأطفال يعانون من نفس المرض في مؤتمر في كارلسروه.[3] ولفغانغ ستوك من جانبه ذكر هذا المرض بحلول سنة 1908.[4] طبيب الأعصاب الألماني هاينريش فوغت هو الآخر كان مهتما بمرض باتن، حيث قام بنشر ورقتين حول هذا الموضوع بين سنتي 1905 و1911. لحقهم فيما بعد الطبيب السويدي تورستن سوجرين، الذي نشر بحلول سنة 1931 وثائق مفصلة حول المرض.[5] كنتيجة لذلك، يسمى مرض باتن أيضا باسم مرض سبيميلير-فوغت-سوجرين-باتن.
بالرغم من أن مرض باتن يعتبر واحدا من أشكال الاضطرابات الليفوسينات السيرويدية العصبية (NCL)، إلا أن بعض الأطباء يستخدمون مصطلح مرض باتن لوصف جميع أشكال الNCL.
العلامات والأعراض
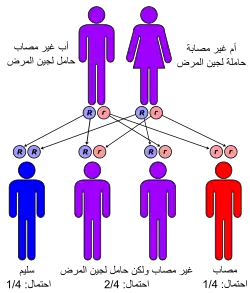
تظهر أعراض داء باتن في وقت مبكر من حياة المريض (في سن 2 إلى 10 سنوات)، يعاني المريض خلالها من اضطرابات في جهازه العصبي، متمثلة في مشاكل تدريجية في الرؤية، أو نوبات صرع. قد تكون العلامات الأولى أيضا عبارة عن تغيرات طفيفة في الشخصية والسلوك، على شكل بطء في التعلم، أو الكلام المتكرر (التلفظ الصدوي)، أو الخرق أو حتى التعثر. قد تشمل الأعراض الأخرى تأخرا في نمو رأس الطفل، وضعف الدورة الدموية في الأطراف السفلية (الساقين والقدمين)، وانخفاض الدهون وكتلة العضلات في الجسم، وانحناء العمود الفقري، وفرط التنفس و/أو نوبات حبس النفس، وطحن الأسنان والإمساك.
بمرور الوقت، يعاني الأطفال المصابون من تفاقم النوبات، والتدهور العقلي بالإضافة الفقدان التدريجي للبصر والكلام والمهارات الحركية. مرض باتن هو مرض قاتل يؤدي في نهاية المطاف إلى موت المصاب. حيث يختلف العمر المتوقع للمريض باختلاف نوع المرض.
تظهر الأعراض الأولى لمرض باتن عند الإناث المصابات بعد عام من وقت ظهورها لدى الذكور، ولكنهن تموتن في المعدل قبل عام من الذكور المصابين.[6]
المسببات
اضطرابات الليفوسينات السيرويدية العصبية هي واحدة من الأمراض المتنحية الجسدية الموروثة، المسؤولة عن غالبية الأمراض العصبية التي تؤثر على الأطفال. يتم في الغالب الإشارة إليها مجتمعة باسم مرض باتن. يحدث مرض باتن حول العالم بوثيرة تقارب شخص واحد لكل 12500 من الساكنة، لكنه يظل أكثر شيوعا في مناطق معينة من العالم. يرتبط كل نوع محدد من مرض باتن بسن ظهور معين وطفرة جينية معنية. المعروف حاليا، هو أن تطور مرض باتن مرتبط بطفرات جينية تصيب 10 جينات.[7]
يحدث مرض باتن عند الأحداث بصفة خاصة، كنتيجة لوجود طفرة في جين CLN3 (المسؤول عن ترميز بروتين الباتينين عند البشر)، [8][9] الذي يقع على مستوى الذراع القصير للكروموسوم رقم 16. بشكل أكثر تحديدا، فإن حوالي 73 في المائة من حالات مرض باتن راجعة بالأساس إلى حذف 1.02 كيلوبايت من هذا الجين (CLN3)، ما يسبب تحولا في الهيكل ينتج عنه جين متحول مبتورة ينتج بروتينا مكونا من 181 حمضا أمينيا فقط بالمقارنة مع الجين السليم الذي ينتج بروتينا يتكون من 438 من الأحماض الأمينية. يشفر جين CLN3 الذي يعمل بشكل طبيعي بروتينا غشائيا كاره للماء يتمركز بشكل رئيسي في الليزوزوم. في المقابل تم العثور على منتج الجين المتحول المكون من 181 حمضا أمينيا في المقام الأول في الشبكة الإندوبلازمية وكذا جهاز جولجي. مع ذلك، تظل الوظيفة الدقيقة لمنتج الجين المتحول غير معروفة.[7]
التشخيص
نظرا لكون داء باتن مرضا نادر الحدوث، فقد يقع المريض ضحية لتشخيص خاطئ، الشئ قد يؤدي إلى زيادة النفقات الطبية والإجهاد الأسري، وإمكانية اتباع أشكال علاج غير صحيحة يمكن أن تؤدي في النهاية إلى تفاقم حالة المريض. لكن مع ذلك، يمكن تشخيص مرض باتن إذا تم اكتشافه بشكل صحيح. ضعف البصر مثلا هو أكثر الأعراض التي يمكن ملاحظتها بشكل واضح للكشف عن هذا المرض. حيث أن أي شك في صحة عين المريض كيفما كاد قد يكون مؤشرا لاصابة الطفل بالمرض.[10] يجب أولا فحص الأطفال أو أي شخص يشتبه في إصابته بمرض باتن من قبل طبيب عيون أو أخصائي نظارات. حيث يتم إجراء فحص قاع العينين، الذي يساعد في الكشف عن تشوهات ضعف الرؤية الشائعة، على غرار دقة طبقة ظهارة الشبكية في البقعة المركزية.[10] على الرغم من أن هذا الفحص يمكن أن يؤشر أيضا لوجود مجموعة متنوعة من الأمراض الأخرى، فينبغي أن ينظر لفقدان الخلايا العينية على أنها إشارة تحذير من مرض باتن. إذا تأكد الاشتباه في مرض باتن، يتم اللجوء إلى إجراء مجموعة متنوعة من الاختبارات الأخرى للمساعدة في تأكيد التشخيص بدقة، من بينها:
- اختبارات الدم أو البول، التي يمكن أن تساعد في كشف الحالات الشادة التي قد تشير إلى مرض باتن. على سبيل المثال، تم العثور على مستويات عالية من الدوليكول في بول العديد من الأفراد المصابين بمرض باتن. في حالة وجود خلايا دم بيضاء مجوفة تحتوي على ثقوب أو تجاويف (تمت ملاحظتها بواسطة تحليل اللطاخة الدموية المجهرية)، إلى جانب نتائج أخرى تؤشر لمرض باتن، فإن كل ذلك يشير إلى وجود مرض باتن في شكله الذي يصيب الأحداث، والذي يحدث كنيجة لوجود طفرات في جين الCLN3.[11]
- أخذ عينات من الجلد أو الأنسجة، عن طريق استخلاص عينة صغيرة من الأنسجة، يتم فحصها تحت المجهر الإلكتروني. الشئ الذي يمكن الأطباء من الكشف عن الرواسب النوعية لمرض باتن. تكون هذه الرواسب شائعة في أنسجة بعينها، على غرار الجلد والعضلات والملتحمة والمستقيم. تعد هذه التقنية التشخيصية مفيدة للغاية، ولكن هناك اختبارات أخرى أكثر موثوقية منها يمكن الاعتماد عليها لتشخيص مرض باتن.[11]
- تخطيط أمواج الدماغ (EEG)، تستخدم هذه التقنية مجسات خاصة ملحقة بفروة رأس الشخص. تعمل هذه المجسات على تسجل الإشارات الكهربائية القادمة من الدماغ، الشئ الذي يسمح للخبراء الطبيين بتحليل أنماط النشاط الكهربائي للدماغ، لمراقبة ما إذا كان المريض يعاني من نوبات.[11]
- الدراسات الكهربائية للعيون، تستخدم كما سبق ذكره، بسبب كون فقدان البصر هي السمة الأكثر شيوعا لمرض باتن. حيث أن الاستجابات البصرية المثارة والتخطيط الكهربائي للشبكية، هي اختبارات فعالة للكشف عن مختلف حالات العين الشائعة المرتبطة بمرض باتن الطفولي.
- التصوير المقطعي (CT) أو التصوير بالرنين المغناطيسي (MRI)، التي تسمح للأطباء بأخد صور تفصيلية للدماغ بشكل أفضل. حيث يتم الاستعانة في تقنية التصوير بالرنين المغناطيسي بمجالات مغناطيسية وموجات راديوية للمساعدة في إنشاء صور مفصلة للدماغ. في حين تستعمل تقنية التصوير المقطعي بغرض إنشاء صورة مفصلة لأنسجة الدماغ وهياكله بالاستعانة بالأشعة السينية وأجهزة الكمبيوتر. يمكن لكلتا التقنيتين المساعدة في عملية الكشف عن مناطق الدماغ المتحللة أو الضامرة لدى الأشخاص الذين يعانون من مرض باتن.[11]
- قياس النشاط الانزيمي الخاص بمرض باتن الذي يساعد في تأكيد تشخيصات بعض الطفرات المختلفة الناتجة.[11]
- تحليل الحمض النوي، الذي يمكن استخدامه هو الآخر للمساعدة في تأكيد تشخيص مرض باتن. عندما تُعرف الطفرة، يمكن أيضًا استخدام تحليل الحمض النووي للكشف عن الناقلات غير المتضررة في هذا الحالة. في حالة ما إذا لم يتم تحديد طفرة عائلية أو إذا كانت الطفرات الشائعة غير موجودة، فالتطورات الجزيئية الحديثة كفيلة بذلك، حيث جعلت من الممكن التعرف على التسلسل الكامل لجميع الجنات المعروفة، الشئ الذي زاد من فرص العثور على الطفرات المسؤولة عن مرض باتن.[11]
العلاج
مرض باتن هو مرض عضال؛ يصعب على جسم المريض التفوق عليه. في المقابل يمكن لعلاج أنزيمي يقوم على عقار السرليبوناز ألفا (المعروف تجاريا باسم "برينورا"، والحاصل على ترخيص إدارة الغذاء والدواء الأمريكية) التقليل من آثار شكل محدد من مرض باتن. البرينورا هو أول علاج معتمد لإبطاء فقدان القدرة على المشي لدى الأطفال المصابين بعمر 3 سنوات فما فوق، والذين يعانون من الداء الليبوفوسيني السيرويدي العصبي من النوع الثاني (CLN2)، المعروف أيضا باسم نقص ثلاثي ببتيديل ببتيداز 1 (TPP1).[12]
الأبحاث
في يونيو 1987، أُطلِقت تجربة سريرية من المرحلة الأولى في كلية وايل كورنيل للطب بجامعة كورنيل لدراسة طريقة العلاج الجيني لعلاج علامات وأعراض الداء الليبوفوسيني السيرويدي العصبي الطفلي الآجل LINCL. يعمل الدواء التجريبي من خلال توصيل ناقل نقل الجينات يسمى AAV2CUhCLN2 إلى الدماغ. على الرغم من أن التجربة ليست مطابقة أو معشاة أو عمياء وتفتقر إلى مجموعة تحكم وهمية / إيحائية متزامنة، فإن تقييم متغير النتيجة الأولية يشير إلى تباطؤ تقدم الداء الليبوفوسيني السيرويدي العصبي الطفلي الآجل LINCL في الأطفال الخاضعين للعلاج.[13]
يعتقد الباحثون أن العجز العصبي الشائع في الداء الليبوفوسيني السيرويدي العصبي اليفعي JNCL يمكن أن يكونسببه فرط نشاط مستقبلات AMPA في المخيخ. لاختبار هذه الفرضية، أعطى الباحثون الأدوية المضادة لـ AMPA للفئران المصابة. أظهرت المهارات الحركية للفئران المصابة تحسّنًا ملحوظًا بعد العلاج المضاد، والذي دعم الفرضية القائلة بأن العجز العصبي في الداء الليبوفوسيني السيرويدي العصبي اليفعي JNCL يرجع إلى مستقبلات AMPA المفرطة النشاط. هذا البحث يمكن أن يساعد في النهاية على تخفيف العجز العصبي للداء الليبوفوسيني السيرويدي العصبي اليفعي JNCL في البشر.[14]
في نوفمبر 2006، بعد الحصول على تصريح من إدارة الغذاء والدواء (FDA)، بدأ جراح الأعصاب ناثان سيلدن وطبيب الأطفال بوب شتاينر وزملاؤه في مستشفى دورنبيشر للأطفال في جامعة أوريغون للصحة والعلوم دراسة سريرية حُقِنت فيها خلايا جذعية عصبية منقاة في دماغ دانيال كيرنر، وهو طفل عمره عام يعاني من مرض باتن، وقد فقد القدرة على المشي والتحدث. كان هذا المريض هو الأول من بين ستة ممن يتلقون حقن منتج للخلايا الجذعية من شركة ستيم سيلز StemCells Inc.، وهي شركة بالو ألتو للتكنولوجيا الحيوية. ويُعتقد أن هذه هي أول عمليات زرع للخلايا الجذعية الجنينية في الدماغ البشري. بحلول أوائل ديسمبر، كان الطفل قد تعافى جيدًا بما يكفي للعودة إلى المنزل، ولوحِظت بعض علامات عودة النطق. كان الهدف الرئيسي للتجارب السريرية في المرحلة الأولى هو التحقق من سلامة عملية الزرع.[15] على العموم، أظهرت بيانات المرحلة الأولى أن جرعات عالية من الخلايا الجذعية العصبية البشرية، التي يتم إجراؤها عن طريق إجراء عملية زرع مباشرة في مواقع متعددة داخل الدماغ، تليها 12 شهرًا من كبت المناعة، كانت مقبولة جيدًا لدى جميع المرضى الستة المسجلين في التجربة. بدا أن الحالات الطبية والعصبية والنفسية العصبية للمرضى، بعد عملية الزرع، مُتسقة مع المسار الطبيعي للمرض. توفي دانييل كيرنر في 20 أغسطس 2009.[16]
في عام 2010، تبرعت شيري وجيم فلوريس بمبلغ مليوني دولار، وهي أكبر هبة تُقدم في تاريخ أبحاث مرض باتن، وساهمت مؤسسة ما بعد تجاوز مرض باتن Beyond Batten Disease Foundation بمبلغ 500000 دولار لإنشاء مختبرات للأطباء الباحثين الإيطاليين: بالابيو وساردييلو وزملاؤهم في معهد جان ودان دنكان للأبحاث العصبية في مستشفى تكساس للأطفال Jan and Dan Duncan Neurological Research Institute of Texas Children’s Hospital.
خلال عام 2011، بدأت أول تجارب سريرية مسيطر عليها مع جامعة روتشستر لعلاج مرض باتن. شملت التجربة 30 مريضًا كانوا يعانون من علامات المرض على أمل إبطاء تقدمه.
في نوفمبر 2013، بدأت كلية ويل الطبية بجامعة كورنيل في جلب المشاركين لدراسة سلامة ناقلات نقل الجينات، الموصوفة على أنها تجربة سلامة وفعالية غير عشوائية. كجزء من تجربة بدأتها جامعة روتشستر في مارس 2014، اختُبِرت ميكوفينولات موفيتيل لتحديد فعاليتها وسلامتها باستخدام ناقل نقل الجينات.
في الأمراض المعقدة كمرض باتن، فإن العلاجات التي تعالج جوانب متعددة من المرض في نفس الوقت لها تأثير أكبر من تلك التي تركز على جانب واحد. "وكتب الباحثون أن "استخدام العديد من استراتيجيات العلاج قد يقدم فوائد إضافية للمرضى الذين يعانون من مرض تنكس عصبي، ولكن يجب مقارنة فوائد هذا النهج بعناية مع الآثار الضارة الإضافية التي قد تجلبها العلاجات الموحدة". لاحظ الفريق الطبي أيضًا أنه "على مدار العقدين الماضيين، عمل العلماء والأطباء في مجتمع مرض باتن على ضمان وجود الأدوات اللازمة لتمكين التقدم نحو علاجات فعّالة بوتيرة غير مسبوقة. وقال الباحثون: "إن التقدم الذي أُحرِز مؤخرًا في أبحاث مرض باتن يمنح الأمل في أن تُتاح قريبًا علاجات فعالة وموجهة"، مشيرين إلى أنّ "مجتمع أبحاث أمراض باتن أصبح نموذجًا على مدى تأثير وفعالية بحوث الأمراض النادرة عند العمل معًا".
انظر أيضا
مراجع
- "Batten Disease Fact Sheet". National Institute of Neurological Disorders and Stroke. مؤرشف من الأصل في 1 مايو 2019. اطلع عليه بتاريخ 02 أغسطس 2018. الوسيط
|CitationClass=تم تجاهله (مساعدة); تحقق من التاريخ في:|تاريخ الوصول=(مساعدة) - F. E. Batten: Cerebral degeneration with symmetrical changes in the maculae in two members of a family. In: Transactions of the Ophthalmological Societies of the United Kingdom, Bd. 23, 1903, S. 386–390
- Stengel‘s Syndrome نسخة محفوظة 01 فبراير 2017 على موقع واي باك مشين.
- W. Stock: Über eine bis jetzt noch nicht beschriebene Form der familiär auftretenden Netzhautdegeneration bei gleichzeitiger Verblödung und über Pigmentdegeneration der Netzhaut. In: Klinische Monatsblätter für Augenheilkunde, Stuttgart, 1908, Bd. 5, S. 225–244
- K. G. T. Sjögren: Die juvenile amaurotische Idiotie. Klinische und erblichkeitsmedizinische Untersuchungen. In: Hereditas, Bd. 14, 1931, S. 197–426
- Cialone J, Adams H, Augustine EF, et al. (May 2012). "Females experience a more severe disease course in Batten disease". Journal of Inherited Metabolic Disease. 35 (3): 549–55. doi:10.1007/s10545-011-9421-6. PMC 3320704. PMID 22167274. الوسيط
|CitationClass=تم تجاهله (مساعدة); no-break space character في|تاريخ=على وضع 4 (مساعدة); تحقق من التاريخ في:|تاريخ=(مساعدة) - Jill M. Weimer; Elizabeth Kriscenski-Perry; Yasser Elshatory; David A. Pearce (2002). "The Neuronal Ceroid Lipofuscinoses: Mutations in Different Proteins Result in Similar Disease". NeuroMolecular Medicine. 1: 111–124. doi:10.1385/nmm:1:2:111. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Rakheja D, Narayan SB, Bennett MJ (September 2007). "Juvenile neuronal ceroid-lipofuscinosis (Batten disease): a brief review and update". Current Molecular Medicine. 7 (6): 603–8. doi:10.2174/156652407781695729. PMID 17896996. الوسيط
|CitationClass=تم تجاهله (مساعدة); no-break space character في|تاريخ=على وضع 10 (مساعدة); تحقق من التاريخ في:|تاريخ=(مساعدة) - Cooper JD (June 2008). "Moving towards therapies for juvenile Batten disease?". Experimental Neurology. 211 (2): 329–31. doi:10.1016/j.expneurol.2008.02.016. PMID 18400221. الوسيط
|CitationClass=تم تجاهله (مساعدة); no-break space character في|تاريخ=على وضع 5 (مساعدة); تحقق من التاريخ في:|تاريخ=(مساعدة) - Ostergaard, John R (2016-08-01). "Juvenile neuronal ceroid lipofuscinosis (Batten disease): current insights". Degenerative Neurological and Neuromuscular Disease (باللغة الإنجليزية). 6. doi:10.2147/DNND.S111967. مؤرشف من الأصل في 04 يوليو 2018. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Noah's Hope - Causes and Symptoms of Batten Disease". www.noahshope.com. مؤرشف من الأصل في 28 مايو 2018. اطلع عليه بتاريخ 03 أغسطس 2018. الوسيط
|CitationClass=تم تجاهله (مساعدة); تحقق من التاريخ في:|تاريخ الوصول=(مساعدة) - "Press Announcements - FDA approves first treatment for a form of Batten disease". www.fda.gov (باللغة الإنجليزية). مؤرشف من الأصل في 23 أبريل 2019. اطلع عليه بتاريخ 03 أغسطس 2018. الوسيط
|CitationClass=تم تجاهله (مساعدة); تحقق من التاريخ في:|تاريخ الوصول=(مساعدة) - Kovács, Attila D.; Pearce, David A. (2008-01-01). "Attenuation of AMPA receptor activity improves motor skills in a mouse model of juvenile Batten disease". Experimental Neurology. 209 (1): 288–291. doi:10.1016/j.expneurol.2007.09.012. PMC 4418195. PMID 17963751. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Worgall S, Sondhi D, Hackett NR, et al. (May 2008). "Treatment of late infantile neuronal ceroid lipofuscinosis by CNS administration of a serotype 2 adeno-associated virus expressing CLN2 cDNA". Human Gene Therapy. 19 (5): 463–74. CiteSeerX = 10.1.1.553.872 10.1.1.553.872. doi:10.1089/hum.2008.022. PMID 18473686. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "A stem cell first at OHSU نسخة محفوظة 2012-02-06 على موقع واي باك مشين." The Portland Tribune, Nov 24, 2006
- "Archived copy". مؤرشف من الأصل في 24 أكتوبر 2015. اطلع عليه بتاريخ 21 سبتمبر 2015. الوسيط
|CitationClass=تم تجاهله (مساعدة)صيانة CS1: الأرشيف كعنوان (link)
 بوابة طب
بوابة طب بوابة علم الوراثة
بوابة علم الوراثة بوابة علم الأحياء الخلوي والجزيئي
بوابة علم الأحياء الخلوي والجزيئي