تنكس واليرياني
تَنَكُّسُ واليريانيّ أو تَنَكُّسُ وولريّ هو عملية تنكس نشطة تنتج عندما يتم قطع أو سحق الألياف العصبية ويتحلل جزء المحور العصبي البعيد عن الإصابة (أي أبعد من جسم الخلية العصبية ).[1] تحدث عملية مرتبطة بالموت مرة أخرى أو تنكس رجعي تُعرف باسم "التنكس الشبيه بالوليريان" في العديد من الأمراض التنكسية العصبية، خاصة تلك التي يكون فيها النقل المحوري ضعيفًا مثل التصلب الوحشي الضموري ومرض الزهايمر.[2] تشير دراسات الثقافة الأولية إلى أن الفشل في توفير كميات كافية من البروتين المحوري الأساسي ثنائي نوكليوتيد الأدنين وأميد النيكوتين(أو اختصارا: NMNAT2 )هو حدث رئيسي.[3][4]
| تنكس واليرياني | |
|---|---|
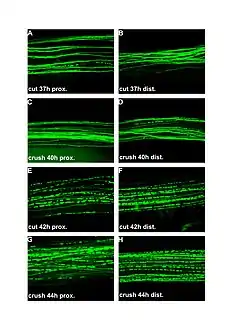 صورة مجهرية فَلْوَرية لتنكس واليرياني صورة مجهرية فَلْوَرية لتنكس واليرياني | |
| معلومات عامة | |
| الاختصاص | طب الجهاز العصبي |
| من أنواع | اضطراب عصبي |
يحدث تنكس واليرياني بعد إصابة محور عصبي في كل من الجهاز العصبي المحيطي (أو اختصارا:PNS) والجهاز العصبي المركزي (أو اختصارا:CNS). يحدث في جزء المحور العصبي البعيد إلى موقع الإصابة ويبدأ عادةً في غضون 24-36 ساعة من الإصابة. يميل القسم البعيد من المحور العصبي إلى البقاء متحمسًا كهربائيًا، قبل الانحطاط. ولكن بعد الإصابة، يتفكك الهيكل العظمي المحوري، ويتفكك الغشاء المحوري. يتبع التنكس المحوري تدهورًا في غمد المايلين وتسلل الضامة. تعمل الضامة، المصحوبة بخلايا شوان، على إزالة الحطام من التنكس.[5][6]
تستجيب خلايا شوان لفقدان المحاور عن طريق بثق أغلفة المايلين، وتقليل تنظيم جينات المايلين، وعدم التمايز والتكاثر. أخيرًا يصطفون في الأنابيب (نطاقات بنجنير) ويعبرون عن جزيئات السطح التي توجه الألياف المتجددة.[7] في غضون 4 أيام من الإصابة، يرسل الطرف البعيد من جزء الألياف العصبية القريب من الإصابة براعم نحو تلك الأنابيب وتنجذب هذه البراعم بواسطة عوامل النمو التي تنتجها خلايا شوان في الأنابيب. إذا وصل البرعم إلى الأنبوب، فإنه ينمو فيه ويتقدم حوالي 1 ملم في اليوم، ويصل في النهاية إلى الأنسجة المستهدفة ويعيدها. إذا لم تتمكن البراعم من الوصول إلى الأنبوب، على سبيل المثال لأن الفجوة واسعة جدًا أو تكون أنسجة ندبة، يمكن أن تساعد الجراحة في توجيه البراعم إلى الأنابيب. التجديد فعال في الجهاز العصبي المحيطي، مع الشفاء شبه الكامل في حالة الآفات التي تحدث بالقرب من طرف العصب البعيد. ومع ذلك، نادراً ما يتم ملاحظة التعافي الكامل في النخاع الشوكي. أحد الاختلافات الجوهرية هو أنه في الجهاز العصبي المركزي، بما في ذلك النخاع الشوكي، يتم إنتاج أغلفة المايلين بواسطة الخلايا قليلة التغصن وليس خلايا شوان.
التاريخ
تم تسمية تنكس واليرياني باسم أوغسطس فولني والر. جرب والر على الضفادع في عام 1850، عن طريق قطع أعصابهم اللسانية البلعومية والأعصاب الموجودة تحت اللسان. ثم لاحظ الأعصاب البعيدة من موقع الإصابة، والتي انفصلت عن أجسامها الخلوية في جذع الدماغ.[5] وصف والر تفكك المايلين، الذي أشار إليه باسم "النخاع"، إلى جزيئات منفصلة بأحجام مختلفة. شكلت المحاور المتدهورة قطرات يمكن أن تكون ملطخة، مما يسمح بإجراء دراسات حول مسار الألياف العصبية الفردية.
تنكس محواري
على الرغم من أن معظم الاستجابات للإصابة تتضمن إشارات تدفق الكالسيوم لتعزيز إعادة سد الأجزاء المقطوعة، فإن الإصابات المحورية تؤدي في البداية إلى تنكس محور عصبي حاد (أو اختصارا:AAD)، وهو الفصل السريع للقريب (الجزء القريب من جسم الخلية) والنهايات البعيدة في غضون 30 دقيقة من الأصابة.[8] بعد الانفصال، تتشكل هياكل المصباح الحثلي في كلا الطرفين ويتم إغلاق الأغشية المقطوعة.[9] تحدث مرحلة الكمون القصيرة في الجزء البعيد والتي تظل خلالها قابلة للإثارة كهربائياً وسليمة هيكلياً.[10] انحطاط يلي مع تورم في غمد المحور العصبي، وأخيرا تشكيل كرات محورية تشبة الخرز. تستغرق العملية 24 ساعة تقريبا في الجهاز العصبي المحيطي، ومدة أطول في الجهاز العصبي المركزي. لا تزال مسارات الإشارات التي تؤدي إلى انحطاط محور الأوكسجين غير مفهومة حاليًا. ومع ذلك، فقد أظهر البحث أن عملية تنكس المحور العصبي (أو اختصارا: AAD) هذه مستقلة عن الكالسيوم.[11]
يحدث التفتت الحبيبي للهيكل الخلوي المحوري والعضيات الداخلية بعد تحلل المحور العصبي. تشمل التغييرات المبكرة تراكم الميتوكوندريا في المناطق الخانقة في موقع الإصابة. تتحلل الشبكة الإندوبلازمية وتتضخم الميتوكوندريا وتتفكك في النهاية. تحدث إزالة البلمرة للأنابيب الدقيقة وسرعان ما يتبعها تدهور في الخيوط العصبية ومكونات الهيكل الخلوي الأخرى. يعتمد التفكك على اليوبيكويتين وبروتينات السيستين (الناجم عن تدفق أيون الكالسيوم)، مما يشير إلى أن التنكس المحوري هو عملية نشطة وليست عملية سلبية كما أسيء فهمها سابقًا.[12] وهكذا يخضع المحور العصبي لتفتيت كامل. معدل التحلل يعتمد على نوع الإصابة كما أنه أبطأ في الجهاز العصبي المركزي منه في الجهاز العصبي المحيطي. هناك عامل آخر يؤثر على معدل التحلل وهو قطر المحور العصبي: تتطلب المحاور الأكبر وقتًا أطول حتى يتحلل الهيكل الخلوي، وبالتالي يستغرق وقتًا أطول حتى يتحلل.
تصفية المايلين
المايلين هو غشاء فسفوليبيد يلتف حول محاور لتزويدها بالعزل. يتم إنتاجه من خلال خلايا شوان في الجهاز العصبي المحيطي، وعن طريق الخلايا قليلة التغصن في الجهاز العصبي المركزي. إزالة الميالين هي الخطوة التالية في تنكس واليرياني بعد التنكس المحوري. يختلف تنظيف بقايا المايلين بالنسبة للجهاز العصبي المحيطي والجهاز العصبي المركزي. الجهاز العصبي المحيطي أسرع وأكثر كفاءة في إزالة حطام المايلين مقارنة بالجهاز العصبي المركزي، وخلايا شوان هي السبب الرئيسي لهذا الاختلاف. جانب رئيسي آخر هو التغيير في نفاذية حاجز الدم والأنسجة في النظامين. في الجهاز العصبي المحيطي (أو اختصارا: PNS)، تزداد النفاذية في جميع أنحاء الجذع البعيد، ولكن تعطيل الحاجز في الجهاز العصبي المركزي يقتصر على موضع الإصابة فقط.[11]
التصفية في الجهاز العصبي المحيطي
استجابة خلايا شوان لإصابة المحور العصبي سريعة. تقدر الفترة الزمنية للاستجابة لتكون قبل بداية تنكس محور عصبي. يعتقد أن النيوريجولين هي المسؤولة عن التنشيط السريع. ينشطون مستقبلات (ErbB2) في خلية زغيبات شوان، مما يؤدي إلى تنشيط بروتين كيناز المنشط بالميتوجين (أو اختصارا:MAPK).[13] على الرغم من ملاحظة نشاط (MAPK)، إلا أن آلية استشعار إصابة خلايا شوان لم يتم فهمها بالكامل بعد. يتبع الاستشعار انخفاض في خلق دهون المايلين ويتوقف في النهاية خلال 48 ساعة. تنفصل أغلفة المايلين عن المحاور في شقوق شميدت-لانترمان أولاً ثم تتدهور بسرعة وتتقلص لتشكل هياكل تشبه الخرز. تستمر خلايا شوان لازالة حطام الميلين التي كتبها المهينة المايلين الخاصة بهم، أن المايلين البلعمي خارج الخلية ويجذب الضامة على حطام الميلين لمزيد من البلعمة.[11] ومع ذلك، فإن الضامة لم تنجذب إلى المنطقة في الأيام القليلة الأولى ؛ ومن ثم فإن خلايا شوان تأخذ الدور الرئيسي في تنظيف المايلين حتى ذلك الحين.
لوحظت خلايا شوان لتجنيد الضامة عن طريق إطلاق السيتوكينات والكيموكينات بعد استشعار إصابة محور عصبي. يساعد تجنيد الضامة في تحسين معدل إزالة حطام المايلين. تطلق البلاعم المقيمة الموجودة في الأعصاب المزيد من الكيموكينات والسيتوكينات لجذب المزيد من الضامة. ينتج العصب المتحلل أيضًا جزيئات التوضيع الكيميائي للبلاعم. مصدر آخر لعوامل توظيف البلاعم هو المصل. لوحظ تأخر تجنيد البلاعم في الفئران التي تعاني من نقص الخلايا البائية التي تفتقر إلى الأجسام المضادة في الدم.[11] تتسبب جزيئات الإشارات هذه معًا في تدفق الضامة، والتي تبلغ ذروتها خلال الأسبوع الثالث بعد الإصابة. بينما تتوسط خلايا شوان المرحلة الأولية لتنظيف حطام المايلين، تأتي البلاعم لإنهاء المهمة. يتم تسهيل البلاعم بواسطة الأ، والتي تُسمى الحطام لإزالتها. تشمل المجموعات الرئيسية الثلاث الموجودة في المصل المكمل والبنتراكسين والأجسام المضادة. ومع ذلك، فقد أظهر المكمل فقط أنه يساعد في بلعمة حطام المايلين.[14]
مورنسون وآخرون (2005)[15] لاحظوا أن خلايا شوان غير الميالين أو النخاعية المتلامسة مع محور عصبي مصاب تدخل دورة الخلية مما يؤدي إلى الانتشار. كانت المدة الزمنية المرصودة لانقسامات خلايا شوان حوالي 3 أيام بعد الإصابة.[16] تُعزى المصادر المحتملة لإشارة الانتشار إلى مستقبلات (ErbB2) ومستقبلات (ErbB3). يمكن أن يؤدي هذا الانتشار إلى زيادة معدلات تنظيف المايلين ويلعب دورًا أساسيًا في تجديد المحاور التي لوحظت في الجهاز العصبي المحيطي. تنبعث خلايا شوان من عوامل النمو التي تجذب براعم محوار جديدة تنمو من الجذع القريب بعد التنكس الكامل للجدعة البعيدة المصابة. هذا يؤدي إلى إعادة تعصيب الخلية أو العضو المستهدف. ومع ذلك، فإن إعادة التعصب ليست بالضرورة مثالية، حيث يحدث تضليل محتمل أثناء إعادة تعصيب المحاور القريبة للخلايا المستهدفة.
التصفية في الجهاز العصبي المركزي
بالمقارنة مع خلايا شوان، تتطلب الخلايا الدبقية قليلة التغصن إشارات محوار للبقاء على قيد الحياة. في مراحل نموها، تخضع الخلايا قليلة التغصن التي تفشل في الاتصال بالمحور وتلقي إشارات المحوار إلى موت الخلايا المبرمج.[17]
أظهرت التجارب التي أجريت على التنكس الواليري أنه عند الإصابة إما أن تخضع الخلايا الدبقية قليلة التغصن لموت مبرمج للخلايا أو تدخل في حالة من الراحة. لذلك، على عكس خلايا شوان، تفشل الخلايا الدبقية قليلة التغصن في تنظيف أغلفة المايلين وحطامها. في التجارب التي أجريت على الفئران،[18] تم العثور على أغلفة المايلين لمدة تصل إلى 22 شهرًا. لذلك، فإن معدلات تطهير غمد المايلين في الجهاز العصبي المركزي بطيئة للغاية ويمكن أن تكون سببًا في إعاقة قدرات التجديد في محاور الجهاز العصبي المركزي حيث لا تتوفر عوامل نمو لجذب المحاور القريبة. ميزة أخرى تنتج في النهاية هي تشكيل الندبة الدبقية. هذا يعيق كذلك فرص التجدد وإعادة التعصب.
تفشل الخلايا قليلة التغصن في تجنيد الضامة لإزالة الحطام. دخول البلاعم بشكل عام إلى موقع الإصابة بالجهاز العصبي المركزي بطيء جدًا. على عكس الجهاز العصبي المحيطي، تلعب الخلايا الدبقية الصغيرة دورًا حيويًا في انحطاط الجهاز العصبي المركزي. ومع ذلك، فإن تجنيدهم أبطأ مقارنة بتجنيد البلاعم في الجهاز العصبي المحيطي(أو اختصارا:PNS) بحوالي 3 أيام. علاوة على ذلك، قد يتم تنشيط الخلايا الدبقية الصغيرة ولكن تتضخم، وتفشل في التحول إلى خلايا بلعمية بالكامل. تلك الخلايا الدبقية الصغيرة التي تتحول بالفعل، تزيل الحطام بشكل فعال. يمكن تحقيق التفريق بين الخلايا الدبقية الصغيرة البلعمية عن طريق اختبار التعبير عن معقد التوافق النسيجي الرئيسي (أو اختصارا:MHC) من الصنف الأول والثاني أثناء تنكس واليريان.[19] معدل الإزالة بطيء للغاية بين الخلايا الدبقية الصغيرة مقارنة بالضامة. يمكن أن يشمل المصدر المحتمل للاختلافات في معدلات الإزالة نقص نشاط الأوبسونين حول الخلايا الدبقية الصغيرة، ونقص النفاذية المتزايدة في الحاجز الدموي الدماغي. يمكن أن يؤدي انخفاض النفاذية إلى إعاقة تسلل البلاعم إلى موقع الإصابة.[11]
أشارت هذه النتائج إلى أن التأخير في تنكس واليريان في الجهاز العصبي المركزي مقارنةً بالجهاز العصبي الحيطي لا يرجع إلى التأخير في التنكس المحوري، بل يرجع إلى الاختلاف في معدلات إزالة المايلين في الجهاز العصبي المركزي والجهاز العصبي المحيطي.[20]
التجدد
التجديد يتبع التنكس. يكون التجدد سريعًا في الجهاز العصبي المحيطي، مما يسمح بمعدلات إعادة نمو تصل إلى 1 ملم في اليوم.[21] قد تكون هناك حاجة أيضًا إلى للسماح بإعادة التعصب المناسب. يتم دعمه بواسطة خلايا شوان من خلال إطلاق عوامل النمو. يكون تجديد الجهاز العصبي المركزي أبطأ بكثير، ويكاد يكون غائبًا في معظم أنواع الفقاريات. قد يكون السبب الرئيسي لذلك هو التأخير في إزالة حطام المايلين. يحتوي حطام المايلين، الموجود في الجهاز العصبي المركزي أو الجهاز العصبي المحيطي، على العديد من العوامل المثبطة. قد يؤدي الوجود المطول لحطام المايلين في الجهاز العصبي المركزي إلى إعاقة التجدد.[22] وجدت تجربة أجريت على نيوتس، الحيوانات التي تتمتع بقدرات سريعة على تجديد محور عصبي للجهاز العصبي المركزي، أن تنكس والريان لإصابة العصب البصري استغرق ما يصل إلى 10 إلى 14 يومًا في المتوسط، مما يشير أيضًا إلى أن الإزالة البطيئة تمنع التجدد.[23]
خلايا شوان والخلايا الليفية العصبية في الجهاز العصبي المحيطي
في الأعصاب السليمة، يُنتج عامل النمو العصبي (أو اختصارا:NGF) بكميات صغيرة جدًا. ومع ذلك، عند الإصابة، يزيد تعبير الرنا المرسل (mRNA) عامل النمو العصبي (NGF) بمقدار خمسة إلى سبعة أضعاف خلال فترة 14 يومًا. تلعب الخلايا الليفية العصبية وخلايا شوان دورًا مهمًا في زيادة التعبير عن NGF mRNA.[24] تعمل البلاعم أيضًا على تحفيز خلايا شوان والأرومات الليفية لإنتاج عامل النمو العصبي(NGF )عبر إنترلوكين 1 المشتق من البلاعم.[25] تشمل الجزيئات العصبية الأخرى التي تنتجها خلايا شوان والخلايا الليفية معًا عامل التغذية العصبية المشتق من الدماغ، وعامل التغذية العصبية المشتق من خط الخلايا الدبقية، وعامل التغذية العصبية الهدبية، وعامل تثبيط اللوكيميا، وعامل النمو الشبيه بالأنسولين، وعامل نمو الأرومة الليفية. تخلق هذه العوامل معًا بيئة مواتية لنمو وتجديد المحور العصبي.[11] بصرف النظر عن عوامل النمو، توفر خلايا شوان أيضًا إرشادات هيكلية لزيادة تعزيز التجديد. خلال مرحلة تكاثرها، تبدأ خلايا شوان في تكوين خط من الخلايا يسمى (نطاقات بنجنير) داخل الأنبوب الصفحي القاعدية. وقد لوحظ أن المحاور تتجدد في ارتباط وثيق بهذه الخلايا.[26] تنظم خلايا شوان إنتاج النينجورين لجزيء التصاق سطح الخلية الذي يعزز النمو.[27] توجه خطوط الخلية هذه تجديد محور عصبي في الاتجاه الصحيح. المصدر المحتمل للخطأ الذي قد ينتج عن ذلك هو عدم تطابق الخلايا المستهدفة كما تمت مناقشته سابقًا.
بسبب عدم وجود عوامل تعزيز مواتية في الجهاز العصبي المركزي، يتم إعاقة التجدد في الجهاز العصبي المركزي.
تنكس واليرياني بطيء
الفئران التي تنتمي إلى السلالة C57BL / Wld s قد أخرت انحلال واليريان،[28] وبالتالي، تسمح بدراسة أدوار أنواع الخلايا المختلفة والعمليات الخلوية والجزيئية الكامنة. أصبح الفهم الحالي للعملية ممكنًا من خلال التجارب على سلالة الفئران Wld s. حدثت الطفرة أولاً في الفئران في Harlan-Olac، وهو مختبر ينتج حيوانات في المملكة المتحدة. طفرة Wld s هي طفرة جسمية سائدة تحدث في كروموسوم الفأر 4.[29][30] الطفرة الجينية عبارة عن تضاعف ترادفي يبلغ 85 كيلو بايت، يحدث بشكل طبيعي. تحتوي المنطقة الطافرة على جينين مرتبطين: نيكوتيناميد أحادي نيوكليوتيد أدينليل ترانسفيراز 1 (أو اختصارا:Nmnat1) وعامل التواجد (Ube4b). تعد منطقة الرابط التي تشفر 18 من الأحماض الأمينية جزءًا من الطفرة.[6] تبين أن التأثير الوقائي لبروتين Wld S يرجع إلى موقع NAD + المركب النشط في منطقة NMNAT1.[31]
على الرغم من أن البروتين الذي تم إنشاؤه يتمركز داخل النواة ولا يمكن اكتشافه بالكاد في المحاور، تشير الدراسات إلى أن تأثيره الوقائي يرجع إلى وجوده في الأجزاء المحورية والنهائية.[32][33] الحماية التي يوفرها بروتين Wld S متأصلة في الخلايا العصبية وليست الخلايا الداعمة المحيطة بها، وهي تحمي المحوار محليًا فقط، مما يشير إلى أن المسار داخل الخلايا مسؤول عن التوسط في انحلال والريان.[34][35]
آثار طفرة Wld S.
الطفرة لا تسبب أي ضرر للماوس. التأثير الوحيد المعروف هو أن تنكس والريان يتأخر لمدة تصل إلى ثلاثة أسابيع في المتوسط بعد إصابة العصب. في البداية، كان يشتبه في أن WLD الصورة تباطؤ الطفرة أسفل تسلل بلعم، ولكن الدراسات الحديثة تشير إلى أن طفرة تحمي المحاور بدلا من تباطؤ الضامة.[6] العملية التي يتم من خلالها تحقيق الحماية المحورية غير مفهومة بشكل جيد. ومع ذلك، تشير الدراسات إلى أن WLD الصورة يؤدي إلى طفرة زيادة النشاط NMNAT1، الذي يؤدي إلى زيادة NAD توليف +.[31] يؤدي هذا بدوره إلى تنشيط العملية المعتمدة على SIRT1 داخل النواة، مما يتسبب في حدوث تغييرات في نسخ الجينات. قد يوفر NAD + في حد ذاته حماية إضافية للمحور عن طريق زيادة موارد طاقة المحور.[36] ومع ذلك، فإن العمل الأحدث يثير الشك في أن NMNAT1 أو NAD + يمكن أن يحل محل جين Wld s كامل الطول.[37] أظهر هؤلاء المؤلفون من خلال الطرق المختبرية والحيوية أن التأثير الوقائي للإفراط في التعبير عن NMNAT1 أو إضافة NAD + لم يحمي المحاور من التنكس. ومع ذلك، أظهرت الدراسات اللاحقة أن NMNAT1 وقائي عند دمجه مع ببتيد استهداف محوري، مما يشير إلى أن مفتاح الحماية التي يوفرها Wld S كان مزيجًا من نشاط NMNAT1 والتوطين المحوري الذي يوفره المجال N-terminal للبروتين الخيمري.[38]
تؤخر الحماية المحورية المقدمة ظهور التنكس الواليري. لذلك يجب تأجيل تنشيط خلية شوان، لأنها لن تكتشف إشارات تدهور محور عصبي من مستقبلات ErbB2. في التجارب التي أُجريت على الفئران المحورة من Wld s، تأخر تسلل البلاعم بشكل كبير لمدة تصل إلى ستة إلى ثمانية أيام.[39] ومع ذلك، بمجرد أن يبدأ التدهور المحوري، يأخذ التنكس مساره الطبيعي، وفيما يتعلق بالجهاز العصبي، يتبع التدهور بالمعدلات الموضحة أعلاه. الآثار المحتملة لهذا البداية المتأخرة هي ضعف قدرات التجدد في الفئران. تشير الدراسات إلى أن التجديد قد يكون ضعيفًا في فئران Wld S، ولكن هذا على الأرجح نتيجة لكون البيئة غير مواتية للتجديد بسبب استمرار وجود الألياف البعيدة غير المتولدة، في حين يتم إزالة الحطام عادةً، مما يفسح المجال لنمو جديد.[40]
جين SARM1
تم إلقاء الضوء بشكل أكبر على مسار تنكس واليرياني من خلال اكتشاف أن عزر وحدة قياس الفا و TIR المعقم المحتوي على بروتين 1 (SARM1) يلعب دورًا مركزيًا في مسار انحلال Wallerian. تم التعرف على الجين لأول مرة في شاشة طفرات ذبابة الفاكهة الصباغية، وبعد ذلك أظهرت الضربات القاضية لمماثلها في الفئران حماية قوية للمحاور المقطوعة مماثلة لتلك الموجودة في Wld S.[41][42]
يحفز SARM1 التوليف والتحلل المائي لـ ADP-ribose دوري (cADPR) من NAD + إلى ADP-ribose.[43] يؤدي تنشيط SARM1 محليًا إلى حدوث انهيار سريع لمستويات NAD + في القسم البعيد من المحور العصبي المصاب، والذي يخضع بعد ذلك للانحطاط.[44] تبين لاحقًا أن هذا الانهيار في مستويات NAD + يرجع إلى أن مجال TIR الخاص بـ SARM1 يحتوي على نشاط انقسام NAD + جوهري.[45] يحتوي بروتين SARM1 على أربعة مجالات، وإشارة توطين الميتوكوندريا، ومنطقة N-terminus المثبطة تلقائيًا والتي تتكون من أشكال أرماديلو / HEAT، وزخارف ألفا معقمة مسؤولة عن التعددية، ومستقبل C-terminus Toll / Interleukin-1 الذي يمتلك نشاطًا إنزيميًا. تنشيط SARM1 كافٍ لانهيار مستويات NAD + وبدء مسار التنكس الواليري.
يساعد نشاط SARM1 في تفسير الطبيعة الوقائية لعامل البقاء NMNAT2، حيث ثبت أن إنزيمات NMNAT تمنع استنفاد NAD + بوساطة SARM1.[46] يتم دعم هذه العلاقة بشكل أكبر من خلال حقيقة أن الفئران التي تفتقر إلى NMNAT2، والتي عادة ما تكون غير قابلة للحياة، يتم إنقاذها بالكامل من خلال حذف SARM1، مما يضع نشاط NMNAT2 في بداية SARM1.[47] تم ربط مسارات إشارات أخرى مؤيدة للانحطاط، مثل مسار MAP kinase، بتنشيط SARM1. لقد ثبت أن إشارات MAPK تعزز فقدان NMNAT2، وبالتالي تعزز تنشيط SARM1، على الرغم من أن تنشيط SARM1 يؤدي أيضًا إلى تشغيل سلسلة MAP kinase، مما يشير إلى وجود شكل من أشكال حلقة التغذية الراجعة.[48][49] أحد التفسيرات للتأثير الوقائي لطفرة Wld S هو أن منطقة NMNAT1، التي عادة ما تكون مترجمة إلى سوما، تحل محل عامل البقاء القابل للتغير NMNAT2 لمنع تنشيط SARM1 عندما تقوم منطقة N-terminal Ube4 من بروتين WldS بترجمة ذلك إلى المحور. حقيقة أن البقاء المعزز لمحاور Wld S يرجع إلى معدل دوران Wld S الأبطأ مقارنةً بـ NMNAT2 يساعد أيضًا في تفسير سبب منح الضربة القاضية SARM1 حماية أطول، حيث سيكون SARM1 غير نشط تمامًا بغض النظر عن نشاط المانع بينما Wld S سوف يتدهور في النهاية. يمكن العثور على الآثار المحتملة لمسار SARM1 فيما يتعلق بصحة الإنسان في النماذج الحيوانية التي تظهر إصابات دماغية مؤلمة، حيث أظهرت الفئران التي تحتوي على عمليات حذف Sarm1 بالإضافة إلى Wld S انخفاض تلف المحور العصبي بعد الإصابة.[50] لقد ربطت طفرات محددة في NMNAT2 آلية تنكس والريان بمرضين عصبيين.
انظر أيضًا
المراجع
- Trauma and Wallerian Degeneration, جامعة كاليفورنيا (سان فرانسيسكو) نسخة محفوظة 2 مايو 2006 على موقع واي باك مشين.
- "Wallerian degeneration, wld(s), and nmnat". Annual Review of Neuroscience. 33 (1): 245–67. 1 June 2010. doi:10.1146/annurev-neuro-060909-153248. PMID 20345246. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Endogenous Nmnat2 is an essential survival factor for maintenance of healthy axons". PLOS Biology. 8 (1): e1000300. January 2010. doi:10.1371/journal.pbio.1000300. PMID 20126265. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "NMNAT: It's an NAD + Synthase… It's a Chaperone… It's a Neuroprotector". Current Opinion in Genetics & Development. 44: 156–162. 2017. doi:10.1016/j.gde.2017.03.014. PMID 28445802. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Experiments on the Section of the Glossopharyngeal and Hypoglossal Nerves of the Frog, and Observations of the Alterations Produced Thereby in the Structure of Their Primitive Fibres". Philosophical Transactions of the Royal Society of London. 140: 423–429. 1 January 1850. doi:10.1098/rstl.1850.0021. JSTOR 108444. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "An 85-kb tandem triplication in the slow Wallerian degeneration (Wlds) mouse". Proceedings of the National Academy of Sciences of the United States of America. 95 (17): 9985–90. August 1998. Bibcode:1998PNAS...95.9985C. doi:10.1073/pnas.95.17.9985. PMID 9707587. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Nerve injury, axonal degeneration and neural regeneration: basic insights". Brain Pathology. 9 (2): 313–25. April 1999. doi:10.1111/j.1750-3639.1999.tb00229.x. PMID 10219748. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "In vivo imaging of axonal degeneration and regeneration in the injured spinal cord". Nature Medicine. 11 (5): 572–7. May 2005. doi:10.1038/nm1229. PMID 15821747. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Endocytotic formation of vesicles and other membranous structures induced by Ca2+ and axolemmal injury". The Journal of Neuroscience. 18 (11): 4029–41. June 1998. doi:10.1523/JNEUROSCI.18-11-04029.1998. PMID 9592084. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Axon degeneration: molecular mechanisms of a self-destruction pathway". The Journal of Cell Biology. 196 (1): 7–18. January 2012. doi:10.1083/jcb.201108111. PMID 22232700. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Why is Wallerian degeneration in the CNS so slow?". Annual Review of Neuroscience. 30 (1): 153–79. 1 July 2007. doi:10.1146/annurev.neuro.30.051606.094354. PMID 17506644. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Multiple forms of Ca-activated protease from rat brain and muscle". The Journal of Biological Chemistry. 259 (5): 3210–8. March 1984. PMID 6321500. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Microanatomy of axon/glial signaling during Wallerian degeneration". The Journal of Neuroscience. 25 (13): 3478–87. March 2005. doi:10.1523/JNEUROSCI.3766-04.2005. PMID 15800203. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Complement depletion reduces macrophage infiltration and ctivation during Wallerian degeneration and axonal regeneration". The Journal of Neuroscience. 18 (17): 6713–22. September 1998. doi:10.1523/JNEUROSCI.18-17-06713.1998. PMID 9712643. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Degeneration of myelinated efferent fibers prompts mitosis in Remak Schwann cells of uninjured C-fiber afferents". The Journal of Neuroscience. 25 (5): 1179–87. February 2005. doi:10.1523/JNEUROSCI.1372-04.2005. PMID 15689554. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Schwann cell properties: 3. C-fos expression, bFGF production, phagocytosis and proliferation during Wallerian degeneration". Journal of Neuropathology and Experimental Neurology. 54 (4): 487–96. July 1995. doi:10.1097/00005072-199507000-00002. PMID 7602323. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Does oligodendrocyte survival depend on axons?". Current Biology. 3 (8): 489–97. August 1993. doi:10.1016/0960-9822(93)90039-Q. PMID 15335686. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Oligodendrocyte survival in Wallerian degeneration". Acta Neuropathologica. 80 (2): 184–91. 31 May 1990. doi:10.1007/BF00308922. PMID 1697140. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "The temporal and spatial activation of microglia in fiber tracts undergoing anterograde and retrograde degeneration following spinal cord lesion". Journal of Neurotrauma. 12 (2): 209–22. April 1995. doi:10.1089/neu.1995.12.209. PMID 7629867. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Delayed macrophage responses and myelin clearance during Wallerian degeneration in the central nervous system: the dorsal radiculotomy model". Experimental Neurology. 129 (2): 225–36. October 1994. doi:10.1006/exnr.1994.1164. PMID 7957737. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Lundy-Ekman, Laurie (2007). Neuroscience: Fundamentals for Rehabilitation (الطبعة 3rd). Saunders. ISBN 978-1-4160-2578-8. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "The Nogo signaling pathway for regeneration block". Annual Review of Neuroscience. 27 (1): 341–68. 21 July 2004. doi:10.1146/annurev.neuro.27.070203.144340. PMID 15217336. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "The early stages of Wallerian degeneration in the severed optic nerve of the newt (Triturus viridescens)". The Anatomical Record. 187 (3): 291–310. March 1977. doi:10.1002/ar.1091870303. PMID 851236. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Changes of nerve growth factor synthesis in nonneuronal cells in response to sciatic nerve transection" (PDF). The Journal of Cell Biology. 104 (6): 1623–31. June 1987. doi:10.1083/jcb.104.6.1623. PMID 3034917. مؤرشف من الأصل (PDF) في 14 أغسطس 2017. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Interleukin 1 increases stability and transcription of mRNA encoding nerve growth factor in cultured rat fibroblasts". The Journal of Biological Chemistry. 263 (31): 16348–51. November 1988. PMID 3263368. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "The degeneration of unmyelinated axons following nerve section: an ultrastructural study". Journal of Neurocytology. 3 (4): 497–512. October 1974. doi:10.1007/BF01098736. PMID 4436692. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Ninjurin, a novel adhesion molecule, is induced by nerve injury and promotes axonal growth". Neuron. 17 (2): 353–61. August 1996. doi:10.1016/S0896-6273(00)80166-X. PMID 8780658. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "The Effectiveness of the Gene Which Slows the Rate of Wallerian Degeneration in C57BL/Ola Mice Declines With Age". The European Journal of Neuroscience. 4 (10): 1000–2. 1 October 1992. doi:10.1111/j.1460-9568.1992.tb00126.x. PMID 12106435. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Perry, V. H., Lunn, E. R., Brown, M. C., Cahusac, S. and Gordon, S. (1990), Evidence that the Rate of Wallerian Degeneration is Controlled by a Single Autosomal Dominant Gene. European Journal of Neuroscience, 2: 408-413. https://doi.org/10.1111/j.1460-9568.1990.tb00433.x نسخة محفوظة 4 يونيو 2018 على موقع واي باك مشين.
- "A gene affecting Wallerian nerve degeneration maps distally on mouse chromosome 4". Proceedings of the National Academy of Sciences of the United States of America. 90 (20): 9717–20. October 1993. Bibcode:1993PNAS...90.9717L. doi:10.1073/pnas.90.20.9717. PMID 8415768. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Increased nuclear NAD biosynthesis and SIRT1 activation prevent axonal degeneration". Science. 305 (5686): 1010–3. August 2004. Bibcode:2004Sci...305.1010A. doi:10.1126/science.1098014. PMID 15310905. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Wallerian degeneration of injured axons and synapses is delayed by a Ube4b/Nmnat chimeric gene". Nature Neuroscience. 4 (12): 1199–206. December 2001. doi:10.1038/nn770. PMID 11770485. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Non-nuclear Wld(S) determines its neuroprotective efficacy for axons and synapses in vivo". The Journal of Neuroscience. 29 (3): 653–68. January 2009. doi:10.1523/JNEUROSCI.3814-08.2009. PMID 19158292. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Prolonged survival of transected nerve fibres in C57BL/Ola mice is an intrinsic characteristic of the axon". Journal of Neurocytology. 22 (5): 311–21. May 1993. doi:10.1007/BF01195555. PMID 8315413. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "The slow Wallerian degeneration gene in vivo protects motor axons but not their cell bodies after avulsion and neonatal axotomy". The European Journal of Neuroscience. 24 (8): 2163–8. October 2006. doi:10.1111/j.1460-9568.2006.05103.x. PMID 17074042. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "A local mechanism mediates NAD-dependent protection of axon degeneration". The Journal of Cell Biology. 170 (3): 349–55. August 2005. doi:10.1083/jcb.200504028. PMID 16043516. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "NAD(+) and axon degeneration revisited: Nmnat1 cannot substitute for Wld(S) to delay Wallerian degeneration". Cell Death and Differentiation. 14 (1): 116–27. January 2007. doi:10.1038/sj.cdd.4401944. PMID 16645633. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Targeting NMNAT1 to axons and synapses transforms its neuroprotective potency in vivo". The Journal of Neuroscience. 30 (40): 13291–304. October 2010. doi:10.1523/JNEUROSCI.1189-10.2010. PMID 20926655. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Genetic influences on cellular reactions to spinal cord injury: activation of macrophages/microglia and astrocytes is delayed in mice carrying a mutation (WldS) that causes delayed Wallerian degeneration". The Journal of Comparative Neurology. 371 (3): 469–84. July 1996. doi:10.1002/(SICI)1096-9861(19960729)371:3<469::AID-CNE9>3.0.CO;2-0. PMID 8842900. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Further studies on motor and sensory nerve regeneration in mice with delayed Wallerian degeneration". The European Journal of Neuroscience. 6 (3): 420–8. March 1994. doi:10.1111/j.1460-9568.1994.tb00285.x. PMID 8019679. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "dSarm/Sarm1 is required for activation of an injury-induced axon death pathway". Science. 337 (6093): 481–4. July 2012. Bibcode:2012Sci...337..481O. doi:10.1126/science.1223899. PMID 22678360. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Sarm1-mediated axon degeneration requires both SAM and TIR interactions". The Journal of Neuroscience. 33 (33): 13569–80. August 2013. doi:10.1523/JNEUROSCI.1197-13.2013. PMID 23946415. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Resolving the topological enigma in Ca 2+ signaling by cyclic ADP-ribose and NAADP". Journal of Biological Chemistry. 294 (52): 19831–19843. 2019. doi:10.1074/jbc.REV119.009635. PMID 31672920. مؤرشف من الأصل في 20 أكتوبر 2020. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "SARM1 activation triggers axon degeneration locally via NAD⁺ destruction". Science. 348 (6233): 453–7. April 2015. Bibcode:2015Sci...348..453G. doi:10.1126/science.1258366. PMID 25908823. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "+ Cleavage Activity that Promotes Pathological Axonal Degeneration". Neuron. 93 (6): 1334–1343.e5. March 2017. doi:10.1016/j.neuron.2017.02.022. PMID 28334607. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "+ depletion". eLife. 5. October 2016. doi:10.7554/eLife.19749. PMID 27735788. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "S, Confers Lifelong Rescue in a Mouse Model of Severe Axonopathy". Cell Reports. 21 (1): 10–16. October 2017. doi:10.1016/j.celrep.2017.09.027. PMID 28978465. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Pathological axonal death through a MAPK cascade that triggers a local energy deficit". Cell. 160 (1–2): 161–76. January 2015. doi:10.1016/j.cell.2014.11.053. PMID 25594179. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "MAPK signaling promotes axonal degeneration by speeding the turnover of the axonal maintenance factor NMNAT2". eLife. 6. January 2017. doi:10.7554/eLife.22540. PMID 28095293. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Attenuated traumatic axonal injury and improved functional outcome after traumatic brain injury in mice lacking Sarm1". Brain. 139 (4): 1094–1105. 2016. doi:10.1093/brain/aww001. PMID 26912636. الوسيط
|CitationClass=تم تجاهله (مساعدة)
 صور وملفات صوتية من كومنز
صور وملفات صوتية من كومنز
 بوابة طب
بوابة طب بوابة علوم عصبية
بوابة علوم عصبية