بريون
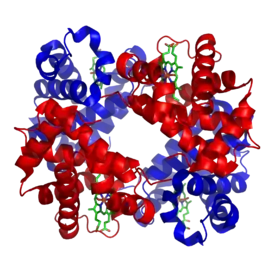
البريون أو الجزيئات البروتينية المسببة للعدوى[1] أو جسيم مُعْدٍ بروتيني[2] (بالإنجليزية: Prion) (اختصار للعبارة ((بالإنجليزية: proteinaceous infectious particle) (![]() i/ˈpriːɒn/[3])) بروتينات معتلة تميّز العديد من الأمراض العصبية المميتة في كلا من الحيوانات والبشر.
لا يزال سبب هذا الاعتلال غير معروف؛ ويشتبه في البنية ثلاثية الأبعاد الغير طبيعية بأنها السبب وراء الخصائص المعدية للبريون.
i/ˈpriːɒn/[3])) بروتينات معتلة تميّز العديد من الأمراض العصبية المميتة في كلا من الحيوانات والبشر.
لا يزال سبب هذا الاعتلال غير معروف؛ ويشتبه في البنية ثلاثية الأبعاد الغير طبيعية بأنها السبب وراء الخصائص المعدية للبريون.
| بريون | |
|---|---|
| معلومات عامة | |
| من أنواع | ممراض ، وبروتين |
يُفترض أن البريونات المكونة من بروتين بريوني هي السبب في اعتلالات الدماغ الإسفنجية المعدية، والتي تشمل قعاص الغنم في الأغنام، والاعتلال الدماغي الإسفنجي البقري في الأبقار (المعروف شعبياً بـ"مرض جنون البقر").
اعتبرت البريونات بشكل فرضي في البشر بأنها السبب في مرض كروتزفيلد جاكوب ومرض كروتزفيلد جاكوب والبديل، داء غيرستمان-ستراوس، و"الأرق الوراثي المميت" ، وداء كورو.
تؤثر جميع أمراض البريون المعروفة في الثدييات على بنية الدماغ أو الأنسجة العصبية الأخرى؛ كما أن جميع هذه الأمراض أمراض مستفحلة، وليس لها علاج فعال معروف، وبالتالي فهي دائماً أمراض قاتلة. الضمور الجهازي المتعدد، وهو مرض تآكل عصبي نادر، يتميز بنسخة معتلة من البروتين تسمى ألفا-ساينوكلين، وبالتالي يمكن تصنيفه أيضًا كأحد أمراض البريون. وقد تم تحديد العديد من بروتينات الخميرة على أنها تحتوي على خصائص بريونية.
يأخذ الدور المفترض للبروتين كعامل معدي موقف النقيض من جميع العوامل المعدية الأخرى المعروفة مثل الفيروسات والبكتيريا والفطريات والطفيليات، فكلها تحتوي على أحماض نووية (ريبوزي، ريبوزي منقوص الأكسجين أو كلاهما). كما أن البريونات الاصطناعية، التي تم إنشاؤها في المختبر بعيدا عن أي مصدر بيولوجي، كانت قدرتها ضئيلة أو منعدمة على إحداث العدوى بأمراض الاعتلال الدماغي الإسفنجي المعدي. ومع ذلك فعندما تم تناول البريونات الاصطناعية مع عامل مرافق، مثل جزيئات الفوسفاتيديتولانيولامين وجزيئات الحمض النووري الريبوزي، فإن قابلية التسبب في هذه الأمراض أصبحت ممكنة.
لا تزال بعض الملاحظات العلمية غير قابلة للتفسير طبقا لفرضية البريون: فمن المعروف أن الفئران التي تعاني من نقص المناعة الوخيم لا تصاب بمرض قعاص الغنم بعد تلقيحها بأنسجة دماغ حيوان مصابة بالمرض، مما يوحي بأنه إما أن دور المناعة في إمراضات البريون غير مفهوم بشكل كامل، وإما أن هناك بعض العيوب الأخرى في الفهم الحالي للفيزيولوجيا المرضية. تبين في الآونة الأخيرة أن مرض قعاص الغنم، وكروتزفيلد - جاكوب قد يتطلبان أحماضًا نووية خاصة بعامل معين لنقل العدوى. ولهذه الأسباب فإن فرضية الاعتلال الدماغي الإسفنجي المعدي/البريون لا تمثل البيانات المرصودة بشكل واف.
أما عن جسيمات البريون فهي مستقرة، وتتراكم في الأنسجة المصابة، وتصاحب تلف الأنسجة وموت الخلايا. ويعني هذا الاستقرار الهيكلي للبريونات أنها تقاوم عملية الإفساد بالمواد الكيميائية والعوامل الفيزيائية، مما يجعل التخلص منها ومنع انتشارها أمرًا صعبًا. تختلف بنية البريون قليلاً بين الأنواع المختلفة، ومع ذلك فإن استنساخ البريون يجعله عرضة للتخلق والانتقاء الطبيعي تمامًا مثل الأشكال الأخرى للاستنساخ.
أمراض البريون المعروفة
الصفات المشتركة لهذه الأمراض
هي اعتلالات اسفنجية عصبية مع فقد عصبي معدية وتغيرات دبقية وترسب لويحات نشوانية وهذه الأمراض غالبا قاتلة ولا يوجد علاج.
علاجه
لا يوجد علاج فعال للأمراض المسببة بالبريون.[4] لم تلق التجارب السريرية على الإنسان أي نجاح، وعرقلتها ندرة الأمراض المسببة بالبريون.[4] على الرغم من أن بعض العلاجات المحتملة تبدو واعدة في المختبر، فإن أيًا منها لم يبد فعالية بمجرد ظهور المرض.[5]
دوره في الأمراض التنكسية العصبية
افتُرض أن إمراضية البريونات والبروتينات المماثلة لها، ظهرت بسبب قدرتها على الرصف الذاتي وما ينتج عن ذلك من نمو متسارع للييفات النشوانية. وثّق بشكل جيد وجود اللييفات النشوانية عند مرضى التنكس العصبي. تظهر هذه الليفات النشوانية كناتج لوجود بروتينات إمراضية تتضاعف بشكل ذاتي وتشكل تكتلات غير وطيفية شديدة الثبات.[6] وعلى الرغم من عدم انطباق ذلك بالضرورة على العلاقة السببية بين النشوان والأمراض التنكسية، إلا أن سمية بعض الأشكال النشوانية وزيادة تشكلها في بعض حالات الأمراض التنكسية العائلية يدعم فكرة أن تشكل النشوان يعد سامًا في العموم.
على نحو خاص، وجد اتحاد للكاظمة تي دي بي-43، وهو بروتين رابط للحمض النووي الريبوزي (RNA)، عند مرضى التصلب الجانبي الضموري، وحدت طفرات في الجينات المرمزة لهذه البروتينات في الحالات العائلية من المرض. عززت هذه الطفرات خلل تطوي البروتينات لتشكل بنى شبيهة بالبريون. تشكل الأشكال مختلة التطوي من الكاظمة تي دي بي-43 شوائب سيتوبلاومية في العصبونات المتضررة، وتستنفذ في النواة. إلى جانب مرض التصلب الجانبي الضموري والتنكس الفصي الجبهي الصدغي، تعد إمراضية الكاظمة تي دي بي-43، خاصية في العديد من حالات مرض ألزهايمر وباركنسون وداء هنتنغتون. يُوجَّه خلل تطوي الكاظمة تي دي بي-43 على نحو كبير بمجالها المشابه للبريون. يميل هذا المجال وراثيًا إلى خلل التطوي، بينما وجد أن الطفرات الإمراضية من الكاظمة تي دي بي-43 تزيد هذا الميل نحو خلل التطوي، ما يفسّر وجود هذه الطفرات في الحالات العائلية من مرض التصلب الجانبي الضموري. أما بالنسبة للفطور، يبدو أن مجال كاظمات تي دي بي-43 المشابهة للبريون ضرورية وكافية لخلل تطوي البروتين وتجمعه.[7]
وبالمثل، حُدّدت الطفرات الإمراضية في المجالات المشابهة للبريون من البروتينات النووية غير المتجامسة hnRNPA2B1 وhnRNPA1 في الحالات العائلية من أمراض التدهور العضلي والدماغي والعظمي وتدهور العصبونات المحركة. يظهر الشكل المثار من جميع هذه البروتينات ميلًا إلى التكتل على ذاته ليشكل لييفات نشوانية، فيما تفاقم هذه الطفرات الإمراضية هذا السلوك وتؤدي إلى تراكم فائض منها.[8]
مراجع
- معني prion في قاموس المعاني نسخة محفوظة 08 سبتمبر 2018 على موقع واي باك مشين.
- عبد الوهاب زايد، ه. ج، هيوز، أ. بورسيدو، ف. نيكولاس، معجم مصطلحات التقنية الحيوية في مجال الأغذية والزراعة، 2006، إصدارات منظمة الفاو، مطبوعات جامعة الإمارات العربية المتحدة، ص268
- "Prion". قاموس أوكسفورد الإنجليزي (الطبعة الثالثة). مطبعة جامعة أكسفورد. سبتمبر 2005. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Aguzzi A, Lakkaraju AKK, Frontzek K (January 2018). "Toward Therapy of Human Prion Diseases" (PDF). Annual Review of Pharmacology and Toxicology. 58: 331–51. doi:10.1146/annurev-pharmtox-010617-052745. PMID 28961066. مؤرشف من الأصل (PDF) في 12 مارس 2020. اطلع عليه بتاريخ أكتوبر 2020. الوسيط
|CitationClass=تم تجاهله (مساعدة); تحقق من التاريخ في:|تاريخ الوصول=(مساعدة) - "Prion Clinic – Drug treatments". مؤرشف من الأصل في 29 يناير 2020. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Eisenberg D, Jucker M (March 2012). "The amyloid state of proteins in human diseases". Cell. 148 (6): 1188–203. doi:10.1016/j.cell.2012.02.022. PMC 3353745. PMID 22424229. الوسيط
|CitationClass=تم تجاهله (مساعدة) - King OD, Gitler AD, Shorter J (June 2012). "The tip of the iceberg: RNA-binding proteins with prion-like domains in neurodegenerative disease". Brain Research. 1462: 61–80. doi:10.1016/j.brainres.2012.01.016. PMC 3372647. PMID 22445064. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Kim HJ, Kim NC, Wang YD, Scarborough EA, Moore J, Diaz Z, MacLea KS, Freibaum B, Li S, Molliex A, Kanagaraj AP, Carter R, Boylan KB, Wojtas AM, Rademakers R, Pinkus JL, Greenberg SA, Trojanowski JQ, Traynor BJ, Smith BN, Topp S, Gkazi AS, Miller J, Shaw CE, Kottlors M, Kirschner J, Pestronk A, Li YR, Ford AF, Gitler AD, Benatar M, King OD, Kimonis VE, Ross ED, Weihl CC, Shorter J, Taylor JP (March 2013). "Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS". Nature. 495 (7442): 467–73. Bibcode:2013Natur.495..467K. doi:10.1038/nature11922. PMC 3756911. PMID 23455423. الوسيط
|CitationClass=تم تجاهله (مساعدة)
 بوابة الكيمياء الحيوية
بوابة الكيمياء الحيوية بوابة طب
بوابة طب بوابة علم الأحياء
بوابة علم الأحياء بوابة علم الأحياء الخلوي والجزيئي
بوابة علم الأحياء الخلوي والجزيئي بوابة علم الفيروسات
بوابة علم الفيروسات بوابة علوم عصبية
بوابة علوم عصبية
 صور وملفات صوتية من كومنز
صور وملفات صوتية من كومنز أنواع من ويكي أنواع.
أنواع من ويكي أنواع.
