هيموجلوبين E
الهيموجلوبين هو هيموجلوبين غير طبيعي بسبب طفرة نقطية في سلسلة بيتا β. في نوكليوتيد 26 وبالتالي هناك تغيير في الحمض الاميني من حمض الجلوتاميك (glutamic acid) إلى لايسين(lysine). الهيموجلوبين E شائع جدا بين الناس من سلالة جنوب شرق آسيا, شرق الهندي, السريلانكية و بنجلاديش .[1][2]
| هيموجلوبين E | |
|---|---|
| معلومات عامة | |
| من أنواع | اعتلال الهيموغلوبين ، وفقر الدم الانحلالي الخلقي |

هذه الطفرة ßE تؤثر على التعبير الجيني ل بيتا β الذي ينتج عنه إنشاء موقع بديل للربط (الوصل المتبادل) في mRNA في الشفرة الجينية أو الكودون 25-27 الخاص ب جين بيتا جلوبين. ينتج عن هذه العملية نقص معتدل في β mRNA الطبيعي وبالتالي إنتاج كميات صغيرة من β mRNA الغير طبيعي. انخفاض بناء سلاسل β جلوبين (beta globin chain) قد يسبب مرض بيتا ثلاسيميا. أيضا هذا الهيموجلوبين البديل(HbE) لديه ضعف في الاتحاد بين سلاسل ألفا وبيتا غلوبين، مما يسبب في عدم الإستقرار عندما يكون هناك كمية عالية من الأكسدة.[3] HbE يمكن الكشف عنه عن طريق الاستشراد الكهربائي أو الهجرة الكهربائية (electrophoresis).
مرض الهيموجلوبين-EE) E)
مرض الهيموجلوبينE هو مرض وراثي ينتج عندما نسل يرث جين HbE من كلا الوالدين. عند الولادة، الرضيع متماثل الزيجوت homozygous بالنسبة ل أليل الهيموجلوبين E لا تظهرعليه أعراض المرض وذلك بسبب وجود HbF (الهيموغلوبين الجنيني). في الأشهر الأولى من الحياة، الهيموغلوبين الجنيني يختفي وكمية الهيموجلوبين E تزيد، لذلك الرضيع تبدأ لديه بيتا ثلاسيميا خفيفة. الأشخاص الذين لديهم مغاير الزيجوت (heterozygote ) لالهيموجلوبين E (واحد طبيعي أليل والاليل الاخر غير طبيعي ) لا تظهر عليهم أية أعراض (عادة لا يعانون من فقر الدم أو انحلال الدم). هناك بعض الحالات المرتبطة بانحلال الدم.[4] الشخص متماثل الزيجوت homozygous بالنسبة لأليل الهيموجلوبين E (كلا الأليلين غير طبيعين) يعانون من فقر دم انحلالي خفيف وتضخم طحال خفيف.
الهيموجلوبين E سمة/ الهيموجلوبين E الصغرى: مغاير الزيجوت (Heterozygous) ل AE )HbE)
يحدث عندما جين الهيموغلوبين E موروثة من أحد الوالدين وجين الهيموغلوبين A من الآخر. يسمى هذا سمة الهيموجلوبين E ، و هو ليس مرضا. الناس الذين لديهم سمة الهيموجلوبينE (مغاير الزيجوت) لايعانون من أعراض ظاهرة وعادة لا ينتج عنه مشاكل صحية. قد يكون لديهم انخفاض حجم الكرية الوسطي (MCV) وخلايا الدم الحمراء تكون غير طبيعية (الخلايا المستهدفة). لها أهمية سريرية محصورة نظرا لإمكانية نقل E أو بيتا-ثلاسيميا.[بحاجة لمصدر]
تجمع مغاير الزيجوت لSE) HbE)
تجمع الزيجوت (Compound heterozygotes) لمرض الهيموجلوبين المنجلي E ينتج عند توريث جين الهيموغلوبين E من أحد الوالدين وجين الهيموغلوبين S من الاخر. كلما انخفضت كمية الهيموغلوبين الجنيني وارتفعت كمية الهيموغلوبين S كلما ظهر فقر دم انحلالي خفيف في مرحلة مبكرة من التنمية.
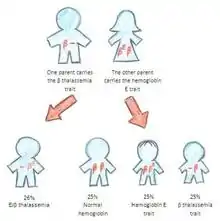
الهيموجلوبين E/بيتا- الثلاسيميا
الاشخاص الذين لديهم الهيموجلوبين E/بيتا-ثلاسيميا هو بسبب وراثة جين الهيموغلوبين E من أحد الوالدين وجين بيتا الثلاسيميا من الاخر. الهيموجلوبين E/بيتا-الثلاسيميا هو مرض حاد، وإلى الان لا يوجد لديه علاج شامل. لأنه يؤثرعلى أكثر من مليون شخص في العالم.[5] عواقب الهيموجلوبين E/بيتا-ثلاسيميا عندما لا تعالج يمكن أن تكوْن فشل القلب, تضخم الكبد, مشاكل في العظام، إلخ.
هناك مجموعة متنوعة من الأنماط الوراثية اعتمادا على تفاعل HbE و ألفا ثلاسيميا. وجود α-الثلاسيميا يقلل من كمية HbE والتي عادة ما تكون موجودة في HbE مغاير الزيجوت. في حالات أخرى، اتحاد طفرات محدده للثلاسيميا، فإنه يوفر زيادة مقاومة الملاريا (المتصورة المنجلية).[6]
علم الأوبئة
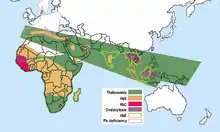
الهيموجلوبين E هو الأكثر انتشارا في البر الرئيسى جنوب شرق آسيا (تايلاند وميانمار وكمبوديا ولاوس وفيتنام[7]) ، حيث انتشاره يمكن أن يصل إلى 30 أو 40% ، شمال شرق الهند، حيث يصل في بعض المناطق معدل ناقلين المرض إلى 60% من السكان. في تايلاند الطفرة يمكن أن تصل إلى 50 أو 70% و هو أعلى في شرق البلاد. في سريلانكا، فإنه يمكن أن تصل إلى 40% ويؤثر على سلالة السنهالية و Vedda.[8][9] كما أنها وجدت بنسبة عالية في بنغلاديش وإندونيسيا.[10][11] و يمكن أن تظهر أيضا في الاشخاص من السلالة التركية، الصينية والفلبينية.[1] ومن المتوقع أن الزيادة في الطفرة حصلت خلال 5000 سنة الماضية.[12] في أوروبا تم العثور على حالات من الأسرالمصابة بالهيموجلوبين E ، ولكن في هذه الحالات، الطفرة تختلف من تلك التي وجدت في جنوب شرق آسيا. وهذا يعني أنه قد تكون هناك أصول مختلفة من طفره ßE .[13][14]
المراجع
- Hemoglobin E Trait - Health Encyclopedia - University of Rochester Medical Center نسخة محفوظة 30 أغسطس 2017 على موقع واي باك مشين.
- (PDF) https://web.archive.org/web/20140624052847/http://www.mhcs.health.nsw.gov.au/publicationsandresources/pdf/publication-pdfs/diseases-and-conditions/9095/oth-9095-eng.pdf. مؤرشف من الأصل (PDF) في 24 يونيو 2014. الوسيط
|CitationClass=تم تجاهله (مساعدة); مفقود أو فارغ|title=(مساعدة) - Chernoff AI, Minnich V, Nanakorn S, et al. (1956). "Studies on hemoglobin E. I. The clinical, hematologic, and genetic characteristics of the hemoglobin E syndromes". J Lab Clin Med. 47 (3): 455–489. PMID 13353880. الوسيط
|CitationClass=تم تجاهله (مساعدة) - A.V. Hoffbrand. Essential Haematology. Second edition. Page 69.
- Vichinsky E (2007). "Hemoglobin E Syndromes". Hematology Am Soc Hematol Educ Program. 2007: 79–83. doi:10.1182/asheducation-2007.1.79. PMID 18024613. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Bachir, D; Galacteros, F (November 2004), Hemoglobin E disease. (PDF), Orphanet Encyclopedia, مؤرشف من الأصل (PDF) في 4 مارس 2016, اطلع عليه بتاريخ 13 يناير 2014 الوسيط
|CitationClass=تم تجاهله (مساعدة); الوسيط|separator=تم تجاهله (مساعدة)CS1 maint: ref=harv (link) - Hemoglobin E Trait, جامعة روتشستر مدرسة الطب وطب الأسنان, مؤرشف من الأصل في 16 ديسمبر 2019, اطلع عليه بتاريخ 13 يناير 2014 الوسيط
|CitationClass=تم تجاهله (مساعدة); الوسيط|separator=تم تجاهله (مساعدة)CS1 maint: ref=harv (link) - Populations of the SAARC Countries: Bio-cultural Perspectives - Google Books نسخة محفوظة 6 يناير 2020 على موقع واي باك مشين.
- (PDF) https://web.archive.org/web/20170331032215/http://php.scripts.psu.edu/nxm2/1985%20Publications/1985-roychoudhury-nei.pdf. مؤرشف من الأصل (PDF) في 31 مارس 2017. الوسيط
|CitationClass=تم تجاهله (مساعدة); مفقود أو فارغ|title=(مساعدة) - Genetic Disorders of the Indian Subcontinent - Google Books نسخة محفوظة 3 يناير 2020 على موقع واي باك مشين.
- Olivieri NF, Pakbaz Z, Vichinsky E (2011). "Hb E/beta-thalassaemia: a common & clinically diverse disorder". Indian J. Med. Res. 134: 522–31. PMC 3237252. PMID 22089616. الوسيط
|CitationClass=تم تجاهله (مساعدة)صيانة CS1: أسماء متعددة: قائمة المؤلفون (link)CS1 maint: Multiple names: authors list (link) - Ohashi; et al. (2004). "Extended linkage disequilibrium surrounding the hemoglobin E variant due to malarial selection". Am J Hum Genet. 74 (6): 1189–1208. doi:10.1086/421330. PMC 1182083. PMID 15114532. الوسيط
|CitationClass=تم تجاهله (مساعدة) Free full text - Kazazian HH, JR., Waber PG, Boehm CD, Lee JI, Antonarakis SE, Fairbanks VF. (1984). "Hemoglobin E in Europeans: Further Evidence for Multiple Origins of the βE-Globin Gene". Am J Hum Genet. 36 (1): 212–217. PMC 1684388. PMID 6198908. الوسيط
|CitationClass=تم تجاهله (مساعدة)صيانة CS1: أسماء متعددة: قائمة المؤلفون (link)CS1 maint: Multiple names: authors list (link) Free full text - Bain, Barbara J (June 2006). Blood cells: a practical guide (الطبعة 4th). Wiley-Blackwell. ISBN 978-1-4051-4265-6. الوسيط
|CitationClass=تم تجاهله (مساعدة)
وصلات خارجية
- الهيموجلوبين E وقائع من واشنطن وزارة الصحة،
- الجمعية الأمريكية لأمراض الدم البرنامج التعليمي الشخصي الهيموجلوبين E اضطرابات
- Orphanet موسوعة دخول الهيموجلوبين E
- الهيموجلوبين E في الأوروبيين
 بوابة علم الأحياء الخلوي والجزيئي
بوابة علم الأحياء الخلوي والجزيئي بوابة طب
بوابة طب بوابة الكيمياء الحيوية
بوابة الكيمياء الحيوية