نقص الجالاكتوز إبيميراس
نقص الجالاكتوز إبيميراس، المعروف أيضا باسم نقص غيل، غالاكتوسيميا الثالث[1] ونقص أودب الجالاكتوز -4 إبيميراس،[2] هو نادر، شكل متنحية مقهورة من الجالاكتوز في الدم يرتبط مع نقص في إبيميراس انزيم غالاكتوز.
نقص إيبيميراز الجالاكتوز | |
|---|---|
| معلومات عامة | |
| من أنواع | وجود الغالاكتوز في الدم ، واضطراب صبغي جسدي متنحي |
الأعراض
أعراض الجلوكتوزيميا النوع الثالث هي واضحة من الولادة، ولكن تختلف في شدة اعتمادا على ما إذا كان شكل المرض المحيطي أو المعمم موجود. قد تشمل الأعراض:[3]
- اليرقان عند الأطفال
- نقص التوتر الطفولي
- ملامح ديسمورفيك
- فقدان السمع الحسي العصبي
- إعاقة النمو
- أوجه القصور الإدراكي
- استنفاد خلايا بوركينجي المخيخ
- فشل المبيض (بوي) و هيبيرغوناديسم الضخامي
- تليف كبدى
- الفشل الكلوي
دراسات النوع الثالث لأعراض الجالاكتوز في الدم هي في معظمها وصفية، وتبقى الآليات المسببة للأمراض الدقيقة غير معروفة. ويرجع ذلك إلى حد كبير إلى عدم وجود نماذج حيوانية وظيفية من الجالاكتوز في الدم الكلاسيكية. التطور الأخير من ذبابة الفاكهة ميلانوغاستر غيل متحولة المعرض أعراض الجالكتوزيميا قد تسفر عن نموذج الحيوان في المستقبل واعدة.
علم الوراثه
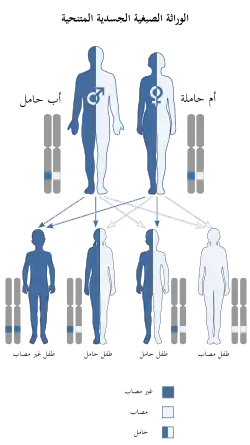
الجالاكتوز إبيميراس نقص هو اضطراب متنحي وراثي،[4] وهو ما يعني أن الجين المعيب يقع على جسيم أوتوسوم، ونسختين من الجين المعيب - واحد من كل الوالدين - مطلوبة لترث هذا الاضطراب. يحمل والدا الفرد المصاب باضطراب جسمي متنحي نسخة واحدة من الجين المعيب، ولكن عادة لا يعاني من أي علامات أو أعراض للاضطراب.
أساس وراثي
وقد تم تحديد الطفرات غيل البشرية المختلفة مما أدى إلى نوع 3 الجالاكتوز في الدم.[5] تحليل وظيفي من هذه الأشكال الإسعافية غيل متحولة تشير إلى أن انخفاض الكفاءة التحفيزية وزيادة احتمال هضم بروتين تعمل بشكل سلبي في النوع الثالث من الجالاكتوز في الدم.
| بقايا متحوله | تأثير البيوكيميائية | المظاهر السريرية |
|---|---|---|
| V94M, K257R, L313M, R335H | ضعف شديد و عدد دوران وخصوصية ثابتة | شديد الجالاكتوز في الدم المعمم.[6] |
| S81R, T150M, P293L | انخفاض عدد دوران معتدل | الجالاكتوز في الدم وسيطة. |
| L183P, D103G, G90E, N34S | ضعف شديد و رقم دوران وخصوصية ثابتة. وزيادة الهضم بروتين. | الجالاكتوز في الدم معمم شديد. |
أساس الكيمياء الحيوية
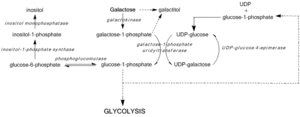
نقص غيل يحول دون تجديد أودب الجلوكوز، ومنع تشكيل الجلوكوز -1 الفوسفات ويؤدي إلى تراكم الجلاكتوز والجلاكتوز -1 الفوسفات. وقد تبين أن مستويات عالية من الجالاكتوز -1 الفوسفات تتداخل مع فوسفهوغلوكوموتاز،[7] فوسفوريلاز الجليكوجين،[8] أودب-غليكوبيروفوسفوريلاز،[9] ونشاط مونوفوسفهاتاز إينوزيتول[10] في النماذج البكتيرية وفي المختبر، ولكن في الجسم الحي سمات آليات لم يتم تأكيدها بعد. بغض النظر عن، متوسط مستويات الجالاكتوز -1 الفوسفات بمثابة التنبؤات الأكثر دقة لشدة الأعراض المرتبطة النوع الثالث من الجالاكتوز في الدم.[11]
انسداد مسار ليلور عن طريق نقص غيل أو اختلال وظيفي ينشط مسارات بديلة من استقلاب الجلوكوز ويؤدي إلى تشكيل غالاكتيتول و غالاكتونات. يتم استقلاب غالاكتونت بواسطة مسار الفوسفات البنتوز، ولا يعتبر سامة.[12] غالاكتيتول، ومع ذلك، قد تتراكم في ألياف العدسة، وتضارب عدسة نفاذية الخلايا الظهارية وتؤدي إلى موت الخلايا وتشكيل الساد.[13] غيل نقص أيضا يتلوى جليكوليبيد والبروتين سكري بسبب انخفاض إنتاج أودب-غالناك من أودب-غلناك.
التشخيص
الفحص للكشف عن مستويات الجالاكتوز مرتفعة قد كشف نقص غيل أو اختلال وظيفي لدى الرضع، ودراسات طفرة ل غيل متاحة سريريا.[14]
تصنيف
هناك نوعان من نقص إبيميراس: نقص حميدة ونقص الكبد الحاد. شكل حاد يشبه الجالاكتوز في الدم.
العلاج
الأفراد الذين يعرضون مع النوع الثالث من الجالاكتوز في الدم يجب أن تستهلك النظام الغذائي اللاكتوز والجلوكتوز مقيدة خالية من منتجات الألبان والنباتات موسيلاجينوس. التقييد الغذائي هو العلاج الحالي الوحيد المتاح لنقص غيل. كما أن البروتين السكري والبروتين السكري الأيض توليد الجالاكتوز الذاتية، ومع ذلك، لا يمكن حل الجالاكتوز في الدم النوع الثالث فقط من خلال تقييد الغذائية.
مراجع
- الوراثة المندلية البشرية عبر الإنترنت (OMIM) Galactose epimerase deficiency -230350
- الوراثة المندلية البشرية عبر الإنترنت (OMIM) UDP-Galactose-4-Epimerase -606953
- "Generalised uridine diphosphate galactose-4-epimerase deficiency". Arch. Dis. Child. 80 (4): 374–6. April 1999. doi:10.1136/adc.80.4.374. PMID 10086948. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "The molecular basis of UDP-galactose-4-epimerase (GALE) deficiency galactosemia in Korean patientsxz". Genetics in Medicine. 7 (9): 646–9. November 2005. doi:10.1097/01.gim.0000194023.27802.2d. PMID 16301867. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Timson DJ (December 2005). "Functional analysis of disease-causing mutations in human UDP-galactose 4-epimerase". FEBS J. 272 (23): 6170–7. doi:10.1111/j.1742-4658.2005.05017.x. PMID 16302980. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Galactose toxicity in animals". IUBMB Life. 61 (11): 1063–74. November 2009. doi:10.1002/iub.262. PMID 19859980. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "The roles of galactitol, galactose-1-phosphate, and phosphoglucomutase in galactose-induced toxicity in Saccharomyces cerevisiae". Biotechnol. Bioeng. 101 (2): 317–26. October 2008. doi:10.1002/bit.21890. PMID 18421797. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Kinetics of purified liver phosphorylase". J. Biol. Chem. 241 (17): 3873–81. September 1966. PMID 5920799. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Overexpression of human UDP-glucose pyrophosphorylase rescues galactose-1-phosphate uridyltransferase-deficient yeast". Biochem. Biophys. Res. Commun. 271 (2): 392–400. May 2000. doi:10.1006/bbrc.2000.2629. PMID 10799308. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Bhat PJ (January 2003). "Galactose-1-phosphate is a regulator of inositol monophosphatase: a fact or a fiction?". Med. Hypotheses. 60 (1): 123–8. doi:10.1016/S0306-9877(02)00347-X. PMID 12450779. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Risk factors for premature ovarian failure in females with galactosemia". J. Pediatr. 137 (6): 833–41. December 2000. doi:10.1067/mpd.2000.109148. PMID 11113841. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Urinary galactonate in patients with galactosemia: quantitation by nuclear magnetic resonance spectroscopy". Pediatr. Res. 42 (6): 855–61. December 1997. doi:10.1203/00006450-199712000-00022. PMID 9396569. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "The effect of an aldose reductase inhibitor on the galactose-exposed rabbit lens". Biochim. Biophys. Acta. 158 (3): 472–5. June 1968. doi:10.1016/0304-4165(68)90305-x. PMID 5660111. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Molecular characterization of a unique patient with epimerase-deficiency galactosaemia". J. Inherit. Metab. Dis. 21 (4): 341–50. June 1998. doi:10.1023/A:1005342306080. PMID 9700591. الوسيط
|CitationClass=تم تجاهله (مساعدة)
روابط خارجيه
 بوابة طب
بوابة طب
