مرض مينكيس
مرض مينكيس (بالإنجليزية: Menkes disease) والمعروف أيضا بمتلازمة مينكيس،[1][2] هو عبارة عن مرض وراثي متنحي مرتبط بالكروموسوم X حيث يؤثر على مستويات النحاس ،[3] مما يؤدي إلى نقص النحاس في الجسم.[4][5] بداية مرض مينكيس عادةً ما تكون أثناء الطفولة، ويصيب حوالي 1 كل 100.000 إلى 200.000 من حديثي الولادة.[6] الأطفال المصابين بمرض مينكيس غالبا لا يعيشون بعد سن 3 سنين. ويكون المرض أكثر شيوعًا في الذكور عن الإناث، لأنه فقط يتطلب نسخة واحدة من الصفة الوراثية المتنحية المرتبطة بالكروموسوم X لكي تظهر الصفة في الذكور ويصاب بالمرض. لكي تصب الإناث بالمرض يحتاجون لوجود نسختين من الصفة الوراثية. يتصف المرض بالشعر المجعد، وفشل في النمو، وتدهور في الجهاز العصبي. وهو ناتج عن حدوث طفرة في الصفة الوراثية المسئولة عن نقل النحاس -ATP7A-، وهي المسؤولة عن صنع بروتين مهم لتنظيم مستويات النحاس في الجسم. وتم وصف المرض في الاصل من قبل جون هانز مينكيس وآخرون في عام 1962.[7]
| مرض مينكيس | |
|---|---|
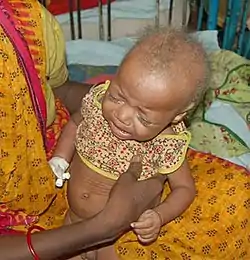 صورة تظهر طفل مريض بمتلازمة مينكيس صورة تظهر طفل مريض بمتلازمة مينكيس | |
| معلومات عامة | |
| الاختصاص | علم الغدد الصم |
| من أنواع | أخطاء خلقية في أيض المعادن ، وشذوذ ساق الشعرة المتلازمي |
أسماء بديلة:
- مرض نقل النحاس
- مرض الشعر الفولاذي
- مرض الشعر المجعد
- متلازمة الشعر المجعد مينكيس
الآلية

تقوم الصفة الوراثية ATP7A بترميز البروتين عبر الغشائي الذي يقوم بنقل النحاس عبر غشاء الخلية. ويوجد في جميع أنحاء الجسم، ماعدا الكبد. في الأمعاء الدقيقة تساعد بروتينات ATP7A في التحكم في امتصاص النحاس من الطعام. وفي الخلايا الأخرى، ينتقل البروتين بين جهاز جولجي وغشاء الخلية للحفاظ على تركيز النحاس في الخلية. يوجد البروتين بشكل طبيعي في جهاز جولجي، الذي يعتبر هامًا لتعديل البروتينات، متضمنًا الإنزيمات. في جهاز جولجي، يوفر بروتين ATP7A النحاس لإنزيمات معينة هامة لبنية ووظيفة العظام، والجلد، والشعر، والأوعية الدموية، والجهاز العصبي.[8] أحد الإنزيمات -ليزيل اوكسيديز بالانجليزية (lysyl oxidase)- يتطلب النحاس للقيام بوظيفته بشكل ملائم. هذا الإنزيم يقوم بربط التروبوكولاجين لإنشاء الياف كولاجين قوية. يساهم الكولاجين التالف في العديد من مظاهر النسيج الضام السابق ذكرها. إذا أصبحت مستويات النحاس مرتفعة، ينتقل البروتين إلى غشاء الخلية لازالة النحاس الزائد من الخلية. الطفرات في الصفة الوراثية ATP7A مثل المسح والإضافة تؤدي إلى مسح أجزاء من الصفة الوراثية، مما يؤدي إلى بروتين ATP7A قصير غير فعال. مما يؤدي إلى خلل في امتصاص النحاس من الطعام ولن يتم إمداد النحاس إلى الانزيمات.
الأعراض
أعراض المرض تتضمن ضعف العضلات، ترهل ملامح الوجه، نوبات، إعاقة ذهنية، وتأخر في النمو. يعاني المرضى من شعر هش واتساع كردوسي. في بعض الحالات النادرة تبدأ الأعراض في وقت متأخر في الطفولة وتكون أقل حدة. الأطفال المصابين قد يولدوا مبكرا. تظهر الأعراض في مرحلة الطفولة غالبًا نتيجة لامتصاص النحاس الغير طبيعي في الامعاء مع النقص الثانوي في إنزيمات الميتوكوندريا التي تعتمد على النحاس. يستمر النمو بشكل طبيعي أو بطئ قليلًا لمدة شهرين إلى ثلاثة شهور ثم يكون هناك تاخر حاد في النمو وفقدان لمهارات النمو المبكرة. يتصف مرض مينكيس أيضا بالنوبات، وفشل النمو، درجة حرارة الجسم اقل من الطبيعي، شعر مجعد لافت للنظر، وعديم اللون، أو لديه لون الفولاذ، وسهل الكسر. يمكن أن يكون هناك تدهور عصبي شديد في المنطقة الرمادية في الدماغ[9] ويمكن أيضا ان تكون الشرايين في الدماغ ملتوية وتكون جدرانها الداخلية مهترئة ومشقوقة. وهذا قد يؤدي إلى تمزق أو انسداد الشرايين. كما قد تؤدي هشاشة العظام إلى الكسور. متلازمة القرن القذالي بالانجليزية (Occipital horn syndrome) هي شكل خفيف من متلازمة مينكيس التي تبدأ في وقت مبكر أو المرحلة الوسطى من الطفولة ويتميز بترسيب املاح الكالسيوم في عظام قاعدة الجمجمة (عظم القذالي)، والشعر الخشن، وجلد ومفاصل سائبة.
السبب والوقاية
الطفرات في الصفة الوراثية ATP7A الموجودة على الكروموسوم Xq21.1، [10] تؤدي إلى مرض مينكيس.[11] ويتم توريث هذا المرض كصفة متنحية مرتبطة بالكروموسوم X.[12] حوالي 30% من حالات مرض مينكيس تنتج عن طفرات بينما 70% تكون موروثة غالبا من الأم.[6] على الرغم من أن المرض أكثر شيوعا في الذكور، الا انه يمكن للإناث ان يصابوا بالمرض. نتيجة طفرة في جين ATP7A، يتم توزيع النحاس بشكل سيئ للخلايا في الجسم. مما يؤدي إلى تراكمه في بعض الأنسجة، مثل الأمعاء الدقيقة والكلى، في حين أن الدماغ والأنسجة الأخرى لديهم مستويات منخفضة بشكل غير عادي. نقص إمداد النحاس يمكن أن يقلل من نشاط العديد من الإنزيمات التي تعتمد على النحاس والتي هي ضرورية لبنية ووظيفة العظام والجلد والشعر والاوعية الدموية والجهاز العصبي مثل اليزيل اوكسيديز.[13] كما هو الحال مع غيرها من الامراض المرتبطة بالكروموسوم X، الأطفال الإناث من أم حاملة لديهم فرص متساوية للاصابة بالمرض، ولكن عادة لا يصابون؛ الأطفال الذكور لديهم فرص متساوية للاصابة بهذا المرض أو عدم الاصابة. قد يقدم استشاري الوراثة نصائح مفيدة.
التشخيص
متلازمة مينكيس يمكن تشخيصها عن طريق اختبارات الدم لمستويات النحاس والسيرولوبلازمين، عينة من الجلد، والفحص المجهري الضوئي للشعر لعرض التغيرات المميزة لمينكيس. يتم عمل أشعة سينية للجمجمة والهيكل العظمي للكشف عن وجود عيوب تكوين العظام.[6] نسبة حمض الهوموفانيلك إلى حمض الفينيل مانديلك (homovanillic acid/vanillylmandelic acid)في البول تم اقتراحها كطريقة للكشف المبكر عن المرض.[14][15] بما أن 70% من الحالات تكون موروثة، فيمكن عمل تحليل وراثي للأم للكشف عن وجود طفرة في جين ATP7A.
العلاج والتنبؤ
لا يوجد شفاء من مرض مينكيس ويبقى التنبؤ بتطور المرض سيئ للغاية. العلاج المبكر بحقن النحاس (في صورة أملاح الاسيتات) قد يكون مفيد إلى حد ما.[16] العلاجات الأخرى تكون للاعراض وداعمة. توجد علاجات للتخفيف من بعض الأعراض تتضمن، مسكنات الالم، علاج النوبات، انبوبة التغذية عند الضرورة، وايضا العلاج الطبيعي.[17]
الإنتشار
دراسة أوروبية واحدة ذكرت معدل 1 لكل 254,000;[18] بينما ذكرت دراسة يابانية معدل 1 كل 357,143.[19] ولا يوجد ارتباط معروف بين هذا المرض والصفات الوراثية الأخرى أو اصل عرقي.
المصادر
- الوراثة المندلية البشرية عبر الإنترنت (OMIM) 309400
- James, William; Berger, Timothy; Elston, Dirk (2005). Andrews' Diseases of the Skin: Clinical Dermatology. (10th ed.). Saunders. صفحة 765. ISBN 0-7216-2921-0. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Menkes syndrome" في معجم دورلاند الطبي
- Vest, Katherine E.; Hashemi, Hayaa F.; Cobine, Paul A. (2013). "Chapter 13 The Copper Metallome in Eukaryotic Cells". In Banci, Lucia (Ed.) (المحرر). Metallomics and the Cell. 12. Springer. doi:10.1007/978-94-007-5561-10_12. ISBN 978-94-007-5560-4. الوسيط
|CitationClass=تم تجاهله (مساعدة)صيانة CS1: نص إضافي: قائمة المحررون (link) electronic-book ISBN 978-94-007-5561-1 ISSN 1559-0836 electronic-ISSN 1868-0402 - de Bie P, Muller P, Wijmenga C, Klomp LW (Nov 2007). "Molecular pathogenesis of Wilson and Menkes disease: correlation of mutations with molecular defects and disease phenotypes". J. Med. Genet. 44 (11): 673–688. doi:10.1136/jmg.2007.052746. PMC 2752173. PMID 17717039. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Research Overview". themenkesfoundation.org. مؤرشف من الأصل في 19 مارس 2019. اطلع عليه بتاريخ 10 ديسمبر 2015. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Menkes JH, Alter M, Steigleder GK, Weakley DR, Sung JH (1962). "A sex-linked recessive disorder with retardation of growth, peculiar hair, and focal cerebral and cerebellar degeneration". Pediatrics. 29: 764–779. PMID 14472668. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "ATP7A gene". Genetics Home Reference. 2015-12-07. مؤرشف من الأصل في 10 ديسمبر 2017. اطلع عليه بتاريخ 10 ديسمبر 2015. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Barnes N, Tsivkovskii R, Tsivkovskaia N, Lutsenko S (2005). "The copper-transporting ATPases, Menkes and Wilson disease proteins, have distinct roles in adult and developing cerebellum". J Biol Chem. 280 (10): 9640–5. doi:10.1074/jbc.M413840200. PMID 15634671. الوسيط
|CitationClass=تم تجاهله (مساعدة) - الوراثة المندلية البشرية عبر الإنترنت (OMIM) 300011
- Voskoboinik I, Camakaris J (2002). "Menkes copper-translocating P-type ATPase (ATPTA): biochemical and cell biology properties, and role in Menkes disease". J Bioenerg Biomembr. 34 (5): 363–71. doi:10.1023/A:1021250003104. PMID 12539963. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Kim BE, Smith K, Meagher CK, Petris MJ (November 2002). "A conditional mutation affecting localization of the Menkes disease copper ATPase. Suppression by copper supplementation". J. Biol. Chem. 277 (46): 44079–84. doi:10.1074/jbc.M208737200. PMID 12221109. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Scheiber, Ivo; Dringen, Ralf; Mercer, Julian F. B. (2013). "Chapter 11. Copper: Effects of Deficiency and Overload". In Astrid Sigel, Helmut Sigel and Roland K. O. Sigel (المحرر). Interrelations between Essential Metal Ions and Human Diseases. 13. Springer. صفحات 359–387. doi:10.1007/978-94-007-7500-8_11. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Matsuo M, Tasaki R, Kodama H, Hamasaki Y (2005). "Screening for Menkes disease using the urine HVA/VMA ratio". J. Inherit. Metab. Dis. 28 (1): 89–93. doi:10.1007/s10545-005-5083-6. PMID 15702409. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Role of optic microscopy for early diagnosis of Menkes disease". ResearchGate. مؤرشف من الأصل في 22 ديسمبر 2015. اطلع عليه بتاريخ 10 ديسمبر 2015. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Kaler SG, Holmes CS, Goldstein DS (February 2008). "Neonatal diagnosis and treatment of Menkes disease". N. Engl. J. Med. 358 (6): 605–14. doi:10.1056/NEJMoa070613. PMC 3477514. PMID 18256395. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Kaler, Stephen G.; Holmes, Courtney S.; Goldstein, David S.; Tang, Jingrong; Godwin, Sarah C.; Donsante, Anthony; Liew, Clarissa J.; Sato, Susumu; Patronas, Nicholas (2008-02-07). "Neonatal Diagnosis and Treatment of Menkes Disease". New England Journal of Medicine. 358 (6): 605–614. doi:10.1056/NEJMoa070613. ISSN 0028-4793. PMC 3477514. PMID 18256395. مؤرشف من الأصل في 13 ديسمبر 2019. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Tønnesen T, Kleijer WJ, Horn N (February 1991). "Incidence of Menkes disease". Hum. Genet. 86 (4): 408–10. doi:10.1007/BF00201846. PMID 1999344. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Gu YH, Kodama H, Shiga K, Nakata S, Yanagawa Y, Ozawa H (2005). "A survey of Japanese patients with Menkes disease from 1990 to 2003: incidence and early signs before typical symptomatic onset, pointing the way to earlier diagnosis". J. Inherit. Metab. Dis. 28 (4): 473–8. doi:10.1007/s10545-005-0473-3. PMID 15902550. الوسيط
|CitationClass=تم تجاهله (مساعدة)
 بوابة طب
بوابة طب
