متلازمة رومانو-وارد
متلازمة رومانو-وارد (بالإنجليزية: Romano–Ward syndrome) هو الخيار الرئيسي لمتلازمة كيو تي الطويلة . هي حالة مرضية تُسبب بحدوث اضطراب النبض الطبيعي للقلب. يُعتبر هذا الاضطراب هو نوع من متلازمة كيو تي الطويلة، حيثُ تؤدي هذه الحالة التي تُصيب القلب إلى زيادة الوقت الذي تحتاجُه عضلة القلب أكثر من المُعتاد لإعادة عملية الشحن ما بين ضربات القلب. وّإذا لم يُعالج، فسيؤدي ذلك إلى حدوث إغماء ونوبة أو الموت المفاجئ بفعل تأثير ضربات القلب العير مُنتظمة.
| متلازمة رومانو-وارد | |
|---|---|
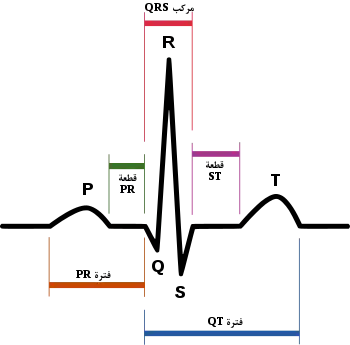 تخطيط كهربائي لقلب طبيعي. تخطيط كهربائي لقلب طبيعي. | |
| معلومات عامة | |
| من أنواع | متلازمة كيو تي الطويلة |
العلامات والأعراض
تزيد متلازمة رومانو وارد من خطورة حدوث اضطرابات في نظم القلب أو عدم انتظام ضربات القلب. عادة ما تكون هذه أحد أشكال تسرع القلب البطيني التي يمكن أن تسبب الإغماء أو النوبات أو حتى الموت المفاجئ.[1] تحدث أيضًا اضطرابات نظم القلب الأقل خطورة مثل الرجفان الأذيني، ما يسبب أعراض تسارع ضربات القلب أو خفقانه. ومع ذلك، فإن العديد من المصابين بمتلازمة رومانو وارد لا يتعرضون إلى عدم انتظام ضربات القلب ويبقون بالتالي لا عرضيين. من المرجح أن تؤدي بعض المواقف إلى حدوث اضطرابات في نظم القلب مثل التمارين الرياضية أو الإجهاد العقلي في النمط الأول لمتلازمة كيو تي الطويلة الخلقية، والضوضاء الصاخبة المفاجئة في النمط الثاني، وأثناء النوم أو فور الاستيقاظ في النمط الثالث.[2]
يمكن تمييز متلازمة رومانو وارد عن الأشكال الأخرى لمتلازمة كيو تي الطويلة من خلال أن تأثير رومانو وارد الوحيد في القلب. بينما ترتبط أشكال أخرى من متلازمة كيو تي الطويلة بالصمم (متلازمة جيرفيل ولانج نيلسن)، والضعف المتقطع والضعف العظمي (النمط السابع متلازمة كيو تي الطويلة، متلازمة أندرسن الطويل)، واضطراب طيف التوحد (النمط الثامن من متلازمة كيو تي الطويلة الخلقية، متلازمة تيموثي)، هذه المظاهر خارج القلب لم تظهر في رومانو وارد.[3]
الأسباب
متلازمة رومانو - وارد هي مصطلح وصفي لمجموعة من الأنواع الفرعية من متلازمة كيو تي الطويلة، وتحديدًا الأنماط 1-6 و9-16.
وصفت عدة أنواع فرعية من متلازمة رومانو وارد بناءً على المتغير الجيني الأساسي. تختلف هذه الأنواع الفرعية في العرض السريري واستجابتها للعلاج. يوجد دليل قوي على أن المتغيرات الجينية المرتبطة بالأنماط الثلاثة الأكثر شيوعًا (النمط الأول والنمط الثاني والنمط الثالث لمتلازمة كيو تي الطويلة) هي بالفعل مسببة للمتلازمة. ومع ذلك، هناك عدم يقين بشأن ما إذا كانت بعض الأنواع الفرعية الأخرى النادرة هي فعلًا مسببة للمرض بنفسها أم أنها تجعل الأفراد أكثر عرضة لإطالة زمن كيو تي استجابةً لعوامل أخرى مثل الأدوية أو انخفاض مستويات البوتاسيوم في الدم (نقص بوتاسيوم الدم).[4]
النمط الأول لمتلازمة كيو تي الطويلة
النمط الأول لمتلازمة كيو تي الطويلة هو النمط الأكثر شيوعًا لمتلازمة رومانو وارد، ويعد مسؤولًا عن 30 إلى 35% من جميع الحالات. عُزل الجين المسؤول -KCNQ1- إلى كروموسوم 11p15.5 ويرمز الوحدة الفرعية ألفا لقناة البوتاسيوم KvLQT1. تتفاعل هذه الوحدة الفرعية مع البروتينات الأخرى (على وجه الخصوص، الوحدة الفرعية minK beta) لإنشاء القناة، والتي تحمل تيار مقوم البوتاسيوم المتأخر IKs المسؤول عن مرحلة إعادة استقطاب جهد القلب.
المتغيرات في KCNQ1 تسبب النمط الأول لمتلازمة رومانو وارد عندما تورث نسخة واحدة من المتغير (وراثة سائدة متغايرة الزويجات). عندما تورث نسختان من المتغير (وراثة متجانسة، وراثة جسمية متنحية)، نكون أمام متلازمة جيرفيل ولانج نيلسن الأكثر شدة، المرتبطة بطول كيو تي بشكل الأكثر وضوحًا، والصمم الحسي الخلقي، وخطر أكبر من عدم انتظام ضربات القلب.
يرتبط النمط الأول بارتفاع مخاطر الإصابة بالإغماء ولكن مع خطر موت مفاجئ أقل من النمط الثاني لمتلازمة كيو تي الطويلة.
قد يؤثر النمط الأول متلازمة كيو تي الطويلة أيضًا على تنظيم الجلوكوز. بعد تناول الجلوكوز، ينتج الأشخاص المصابون بهذا النمط أنسولين أكثر مما هو متوقع، يلي هذه المرحلة فترة من مقاومة الأنسولين. عندما تنخفض المقاومة، يلاحظ أحيانًا انخفاض غير طبيعي في مستويات الجلوكوز في الدم (نقص سكر الدم).[5]
النمط الثاني من متلازمة كيو تي الطويلة
النمط الثاني هو ثاني أكثر أشكال متلازمة رومانو وارد شيوعًا، وهو مسؤول عن 25 إلى 30% من جميع الحالات. ينتج هذا الشكل من متلازمة رومانو وارد عن متغيرات في جين KCNH2 على الكروموسوم 7. KCNH2 (المعروف أيضًا باسم hERG) يغلق قناة البوتاسيوم التي تحمل تيار المعدل الداخلي السريع IKr. يساهم هذا التيار في مرحلة إعادة الاستقطاب النهائي لإمكانات عمل القلب، وبالتالي طول فترة كيو تي.
العلاج
يلعب اختلال التوازن بين الجانبين الأيمن والأيسر لالجهاز العصبي الودي في الإصابة بهذه المتلازمة. بالإمكان التخلص بشكلً مؤقت من هذا الاختلال بعمل إحصار للعقدة النجمية اليسرى وذلك لتقصير فترة كيو تي، وإذا تمت بنجاح يُصبح بالإمكان أجراء عملية استئصال العقدة وتعتبر كعلاج دائم لهذه المتلازمة.[6]
المراجع
- Tester, David J.; Schwartz, Peter J.; Ackerman, Michael J. (2013). Gussak, Ihor; Antzelevitch, Charles (المحررون). Congenital Long QT Syndrome. Electrical Diseases of the Heart: Volume 1: Basic Foundations and Primary Electrical Diseases (باللغة الإنجليزية). Springer London. صفحات 439–468. doi:10.1007/978-1-4471-4881-4_27. ISBN 9781447148814. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Nakajima T, Kaneko Y, Kurabayashi M (2015). "Unveiling specific triggers and precipitating factors for fatal cardiac events in inherited arrhythmia syndromes". Circulation Journal. 79 (6): 1185–92. doi:10.1253/circj.CJ-15-0322. PMID 25925977. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Priori, S. G.; Blomström-Lundqvist, C.; Mazzanti, A.; Blom, N.; Borggrefe, M.; Camm, J.; Elliott, P. M.; Fitzsimons, D.; Hatala, R.; Hindricks, G.; Kirchhof, P.; Kjeldsen, K.; Kuck, K. H.; Hernandez-Madrid, A.; Nikolaou, N.; Norekvål, T. M.; Spaulding, C.; Van Veldhuisen, D. J.; Task Force for the Management of Patients with Ventricular Arrhythmias the Prevention of Sudden Cardiac Death of the European Society of Cardiology (ESC) (29 August 2015). "2015 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death". Europace (باللغة الإنجليزية). 17 (11): 1601–87. doi:10.1093/europace/euv319. ISSN 1099-5129. PMID 26318695. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Giudicessi, John R.; Wilde, Arthur A. M.; Ackerman, Michael J. (October 2018). "The genetic architecture of long QT syndrome: A critical reappraisal". Trends in Cardiovascular Medicine. 28 (7): 453–464. doi:10.1016/j.tcm.2018.03.003. ISSN 1873-2615. PMC 6590899. PMID 29661707. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Demirbilek H, Galcheva S, Vuralli D, Al-Khawaga S, Hussain K (May 2019). "Ion Transporters, Channelopathies, and Glucose Disorders". Int J Mol Sci. 20 (10): 2590. doi:10.3390/ijms20102590. PMC 6566632. PMID 31137773. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Hines, Roberta, Soeltin's Anesthesia and Co-existing Disease (الطبعة 4), Elsevir, صفحة 89 الوسيط
|CitationClass=تم تجاهله (مساعدة); الوسيط|separator=تم تجاهله (مساعدة)CS1 maint: ref=harv (link)
 بوابة طب
بوابة طب