متلازمة برادر- فيلي
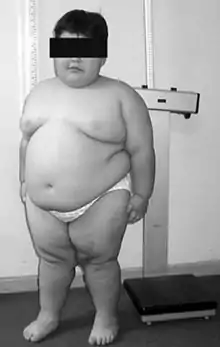
متلازمة برادر- ويلي (بالإنجليزية: Prader-Willi syndrome .. PWS) هي اضطراب جيني نادر حيث يتم حذف أو عدم ترجمة سبع جينات على الكروموسوم 15 (اس 11-13) من الكروموسوم الأبوي (حذف جزئي للكروموسوم15). تم وصفها للمرة الأولى من قبل أندريا برادر (1915-2001)، هينريتش ويلي (1900-1971)، أليكسيس لابهارت (1916-1994)، أندرو زيجلر و جويدو فانكوني عام 1956 في المؤتمر الدولي الثامن لطب الأطفال في عاصمة الدانمارك-كوبنهاغن.[3] خصائص المتلازمة هي:"نقص التوتر العضلي، قصر القامة، عدم اكتمال النمو الجنسي، صعوبات إدراكية، مشاكل سلوكية، و شعور مزمن بالجوع يؤدي إلى الأكل المفرط وسمنة مهددة للحياة".[3] نسبة حدوث المتلازمة 1 لكل 25.000 و 1 لكل 10.000 ولادة. تأتي أهمية المادة المادة الوراثية الأبوية المتأثرة في هذه المتلازمة إلى أن المنطقة المحددة على الكروموسوم .parent-of-origin imprinting المسؤولة عرضة ل مما يعني أنه من مجموعة الجينات الموجودة في تلك المنطقة نسخة واحدة فقط يتم إظهارها والنسخة الأخرى يتم كتمها عن طريق التعلم بالطبع imprinting. ففي حالة متلازمة برادر- ويلي فإن النسخة الأموية من الجينات خامدة (لايتم ترجمتها)، بينما النسخة الأبوية المتغيرة تصبح غير قادرة على العمل (لا يتم ترجمتها أيضا). وهذا يعني أن الناس الطبيعين يملكون نسخة أموية خامدة وأبوية تعمل، بينما الناس المصابين بهذه المتلازمة يملكون نسخة خامدة وأخرى لا تعمل.[3] إن كانت النسخة الأموية لنفس المنطقة هي المتضررة فإن المتلازمة الشقيقة لمتلازمة برادر-ويلي ستظهر وهي متلازمة أنجلمان. مع الفوائد الأخيرة للتشخيص المبكر والتدخلات المستمرة، فإن نسبة السمنة بين الأطفال مع متلازمة برادر-ويلي قد تناقصت لتكون مشابهة للسكان النموذجيين. ومع العلاج السلوكي والعلاجات الاخرى، فإنه يمكن تخفيض تأثيرات المتلازمة.[4][5]
| Prader-Willi syndrome | |
|---|---|
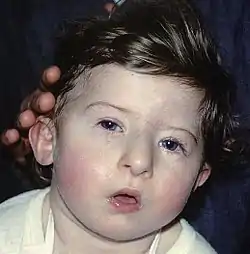 الأشخاص الذين يعانون من متلازمة برادر-ويلي يملكون صفات وجهية مميزة وهي: وجه طويل،صدغ ضيق، شفة عليا رفيعة، وأنف بارز. الأشخاص الذين يعانون من متلازمة برادر-ويلي يملكون صفات وجهية مميزة وهي: وجه طويل،صدغ ضيق، شفة عليا رفيعة، وأنف بارز. | |
| تسميات أخرى | Labhart-Willi syndrome, Prader's syndrome, Prader-Labhart-Willi-Fanconi syndrome [1] |
| النطق | /ˈprɑːdər ˈvɪli/ |
| معلومات عامة | |
| الاختصاص | علم الوراثة الطبية ، وطب الأطفال ، وطب الجهاز العصبي |
| من أنواع | متلازمة ، وسمنة متلازمية |
| التاريخ | |
| سُمي باسم | هاينريش ويلي [2] |
العلامات والأعراض
هنالك العديد من العلامات والأعراض لمتلازمة برادر-ويلي. الأعراض تتراوح من ضعف العضلات في سن الرضاع إلى مشكلات سلوكية في مرحلة الطفولة المبكرة. من الأعراض التي تظهر في سن الرضاع بالإضافة إلى ضعف العضلات، هي غياب التنسيق بين العينين، والبعض قد يولدون بعينين بشكل اللوز، وبسبب ضعف العضلات لدى الرضيع فإنه لا يملك منعكس مص قوي. بكاؤهم ضعيف، ومن الصعب أيقاضهم. وعلامة أخرى انهم يملكون شفة عليا رفيعة.[6] هولم واخرين (1993) قاموا بوصف العلامات والسمات كمؤشرات قبل الاختبار لمتلازمة برادر-ويلي، وإن لم تكن جميعها ظاهرة.
قبل الولادة في داخل الرحم
- انخفاض حركة الجنين
- تكرار أوضاع الجنين غير طبيعية
- موه السلى العرضي ( سائل سلوي زائد)
- غالبا الولادة تكون مقعدية أو قيصرية
- نوام
- نقص التوتر
- صعوبة التغذية (بسبب ضعف توتر العضلات التي تؤثر على منعكس المص)
- صعوبة بدء التنفس
- قصور الغدد التناسلية
مرحلة الطفولة
- تأخر العلامات/التأخر الفكري
- النوم المفرط
- حول العينان
- جنف (غالبا لا يكشف عند الولادة)
- اختفاء الخصية
- تأخر النطق
- ضعف التنسيق البدني
- فرط الأكل، تبدأ ما بين 2 إلى 8 سنوات وتستمر طوال سن البلوغ. لاحظ التغير من صعوبة الأكل في فترة الرضاعة.
- زيادة الوزن المفرط
- اضطرابات النوم
- تأخر سن البلوغ
- قصر القامة
- سمنة
- مرونة مفرطة
سن البلوغ
- العقم (الذكور والإناث)
- قصور الغدد التناسلية
- تناثر شعر العانة
- سمنة
- نقص التوتر (ضعف العضلات)
- صعوبات التعلم/ الوظائف الفكرية محدودة (ولكن بعض الحالات ذات ذكاء متوسط)
- عرضة للإصابة بالسكري
- مرونة مفرطة
المظهر الجسماني
- بروز جسر الأنف
- ضغر الأيدي والأقدام مع تضاؤل الأصابع
- بشرة لينة، سهلة الكدمات
- دهون زائدة، خاصة في الجزء المركزي من الجسم
- جبهة عالية وضيقة
- الشفة العليا رفيعة
- الفم منحن لأسفل
- عينان بشكل اللوز
- لون الشعر والبشرة فاتح مقارنة ببقية أفراد العائلة
- عدم اكتمال التطور الجنسي
- Frequent skin picking
- Striae سطور
- تأخر النمو الحركي
الأعصاب - الادراك
الأفراد الذين يعانون من متلازمة برادر-ويلي هم عرضة لصعوبات التعلم والانتباه. Curfs و Fryns (1992) قاموا بإجراء بحث حول تفاوت درجات عجز التعلم في متلازمة برادر-ويلي.[7] باستخدام مقياس نسبة الذكاء IQ نتائجهم كانت كالتالي:
- 5%: IQ أعلى من 85 (ذكاء متوسط من أعلى إلى أدنى )
- 27%: IQ من 70-85 (عمل فكري بحدود دنيا)
- 39%: IQ 50-70 (عجز فكري منخفض)
- 27%: IQ 35-50 (عجز فكري متوسط)
- 1%: IQ 20-35 (عجز فكري حاد)
- <1%: IQ<20 ( عجز فكري عميق)
Cassidy وجد أنه 40% من الأشخاص الذين يعانون من متلازمة برادر-ويلي يملكون ذكاء بحدود دنيا/ ذكاء متوسط منخفض,[8] وهو رقم أكبر من 32% الموجود في دراسة Curf و Fryn.[7] مع ذلك، أشارت الدراستين إلى أن معظم الأفراد (50-65%) يقعون في مدى الذكاء المتوسط الخفيف/الحد أدنى/ المنخفض. تقارير الأهالي تبين أن بعض الأطفال لديهم IQs >110 ووظائفهم طبيعية في المدرسة.[citation needed] الأطفال الذين يعانون من المتلازمة يظهرون سجل إدراكي غير طبيعي. بالعادة هم أقوياء في نواحي التمييز البصري و الإدراك، بما في ذلك القراءة والمفردات، لكن لغتهم المنطوقة ( أحيانا تتأثر بسبب الخنة المفرطة) إجمالا أضعف من فهمهم. وتم ملاحظة مهارتهم في استكمال أحجية الصور المقطوعة،[9][10] ولكن هذا الأثر قد يكون بسبب ازدياد التدريب.[11]
معالجة المعلومات السمعية والمعالجة التتابعية ضعيفتان نسبيا، وكذلك الحساب ومهارات الكتابة، الذاكرة البصرية والسمعية قصيرة المدى و فترة الانتياه السمعي قد تزداد بتقدم العمر، ولكن القصور في هذه المناطق يستمر طوال سن البلوغ.[9]
السلوك
متلازمة برادر-ويلي عادة تكون مصحوبة مع شهية مفرطة ونهم، وغالبا ما ينتج عنها سمنة مرضية. هذه المتلازمة هي السبب الجيني الأكثر شيوعا للسمنة المرضية عند الأطفال.[12] للان لا يوجد إجماع حول سبب هذا العرض، على الرغم أن التغيرات الجينية على كروموسوم 15 تعطل سير الوظائف الطبيعي للوطاء.[13] من المعلوم أن النواة الوطائية المقوسة تنظم العديد من العمليات الأساسية، بما فيه الشهية، فقد تكون هنالك صلة. عند الأشخاص المصابين بالمتلازمة، الخلايا العصبية التي تنتج هرمون الأوكسايتوسين في الوطاء، وهو هرمون يعتقد أنه يساهم في الشبع، وجدت بأنها غير طبيعية. لديهم مستويات جريلين عالية، والتي يعتقد أنها تؤثر مباشرة على ازدياد الشهية، فرط الأكل، والسمنة .الأشخاص المصابون بمتلازمة برادر-ويلي نص على ضرورة وجود تصور واضح للتوقعات السلوكية، تعزيز المحددات السلوكية وتأسيس روتين منتظم.Cassidy المشاكل النفسية الرئيسية التي يعاني منها الأشخاص مع متلازمة برادر-ويلي تشمل على السلوك القهري ( تتجلى عادة skin picking) و قلق.[14] الأعراض النفسية مثل : الهلوسات، ذهان كبريائي و إحباط، تم وصفها في بعض الحالات [9][15] وتؤثر تقريبا على 5-10% من الشباب.[12] المشاكل النفسية والسلوكية هي الأسباب الأكثر شيوعا للإدخال إلى المستشفى.[16]
الغدد الصم
هنالك العديد من النواحي في متلازمة برادر-ويلي التي تدعم فكرة نقص هرمون النمو في الأشخاص الذين يعانون من المتلازمة. تحديدا، قصر القامة، السمنة مع بنية الجسم غير الطبيعية، انخفاض الكتلة الحرة من الدهون (FFM)، وانخفاض كتلة الجسم الغث (LBM) والمجموع الكلي لإنفاق الطاقة، وانخفاض كثافة العظم. تتميز المتلازمة بقصور الغدد التناسلية. يظهر ذلك بخصية غير نازلة عند الذكور وعنفوان التكظر الباكر الحميد عند الإناث. الخصيتان من الممكن أن ينزلا مع الوقت أو تعالج عن طريق الجراحة أو بديل التستوستيرون. يمكن معالجة عنفوان التكظر عن طريق العلاج بالهرمونات البديلة.
طب العيون
PWS يترافق عادة مع تطور الحول. في واحدة من الدراسات،[17] أكثر من 50% من المرضى لديهم حول، يكون غالبا حول إنسيّ.
الوراثة

تنتج متلازمة برادر ويلي من حذف النسخة الأبوية من الجينين SNRPN و necdin بالإضافة إلى مجموعات من snoRNA وهي: SNORD64,SNORD107.SNORD108 ونسختين منSNORD109 و 29 نسخة منSNORD116 (HBII-85) و 48 نسخة من SNORD115 (HBII52). توجد هذه الجينات جميعها على الكروموسوم 15 في المنطقة 15q11-13.[18][19][20] لذا فإن هذه المنطقة تسمى بمنطقة PWS/AS (منطقة متلازمة برادر-ويلي/متلازمة اْنجلمان)، وهذه المنطقة قد تفقد بواحدة من الاْليات الوراثية العدة، في معظم الحالات تحدث نتيجة احتمالية الطفرات. الاْليات الأخرى الأقل شيوعا تضم: uniparentaldisomy، الطفرات الفرادية، الإزفاء، وحذف الجينات. بسبب التعلم بالطبع، فإن النسخ الأموية من هذه الجينات خامدة، فقط النسخ الأبوية من هذه الجينات يتم تعبيرها.[21][22] متلازمة برادر-ويلي تنتج عن حذف النسخ الأبوية من هذه المنطقة. حذف النسخ الأموية من نفس المنطقة ينتج عنها متلازمة انجلمان. كلا المتلازمتان؛ متلازمة برادر-ويلي وانجلمان تمثلان أولى الحالات المسجلة للاضطرابات عند البشر الناجمة عن التعلم بالطبع. إن خطورة أن يكون إخوة الأطفال المتضررين لديهم المتلازمة أيضا تعتمد على الآلية التي سببت المتلازمة. نسبة أن يكون الإخوة متضررين أيضا تكون أقل من 1% إن كانت الالية التي سببت المتلازمة حذف الجينات أو uniparentaldisomy، وبنسبة تصل إلى 50% إن كانت الية ظهور المتلازمة عند الأطفال المتضررين طفرة في المنطقة المسؤولة عن التعلم بالطبع، ونسبة 25% إذا كان السبب إزفاء أبوي. الفحص السابق للولادة ممكن لتحديد أي من الاليات الجينية المسببة.
في العائلة الواحدة تم اسبعاد أن يكون الحذف الصغير لـ snoRNA HBII-52 في أن يكون مسبب رئيس للمرض.[23] الدراسات على كل من الفئران والبشرأظهرت أن حذف 29 نسخة من C/D box snoRNA SNORD116 (HBII-85) بأنه السبب الرئيس لمتلازمة برادر-ويلي.[24][25][26][27][28]
التشخيص
تؤثر متلازمة برادر ويلي تقريبا على 1 لكل 10.000 إلى 1 لكل 25.000 من حديثي الولادة .[29] وهناك أكثر من 400.000 شخص حول العالم يعيش مع المتلازمة.[30] وتتميز بنقص التوتر، قصر القامة، فرط الأكل، سمنة، وقضايا سلوكية (خصوصا السلوكات المشابهة للاضطراب الوسواسي القهري) ، أيدي وأقدام صغيرة، قصور الغدد التناسلية، وإعاقة فكرية متوسطة.[29] بالرغم من ذلك، مع التشخيص والعلاج المبكر (مثل المعالجة بهرمون النمو)، توقعات سير المرض للأشخاص مع المتلازمة بدأت بالتغير. مثل الذاتوية، متلازمة برادر-ويلي هي اضطراب طيفي و الأعراض قد تتراوح من معتدلة إلى حادة وقد تتغير على مدى حياة الشخص المصاب. وتؤثر المتلازمة على أجهزة الجسم المختلفة. تقليديا، متلازمة برادر ويلي كانت تشخص من خلال الأعراض السريرية. حاليا، المتلازمة تشخص عن طريق الفحص الجيني؛ ينصح بالفحص الجيني لحديثي الولادة مع قصر توتر واضح. التشخيص المبكر للمتلازمة يتيح التدخل المبكر والوصف المبكر لهرمون النمو. حقن هرمون النمو المأشوب يوميا حددت للأطفال مع المتلازمة. هرمون النمو يدعم النمو الخطي ويزيد من كتلة العضلات، وقد يقلل من الانهماك في الأكل وازدياد الوزن. الفحص الجيني هو عماد التشخيص، تحديدا فحص مثيلة الحمض النووي DNA للكشف عن غياب المنطقة الأبوية المساهمة في متلازمة برادر-ويلي/ متلازمة انجلمان (PWS/AS) على الكروموسوم 15q11-q13. هذا الفحص يكشف 97% من الحالات. فحص المثيلة الخاص مهم لتأكيد التشخيص لجميع الأشخاص المصابين ولكن خصوصا لؤلائك الأصغر من إظهار علامات كافية ليتم التشخيص سريريا أو لؤلائك الذين لديهم نتائج لانمطية(شاذة). متلازمة برادر-ويلي في كثير من الأحيان تشخص خطأ كغيرها من المتلازمات لعدم إلمام الكثيرين بها من المجتمع الطبي.[13] أحيانا تشخص خطأ على أنها متلازمة داون, وذلك لأن متلازمة داون نسبيا أكثر تكرارا مقارنة بمتلازمة برادر-ويلي.[13]
العلاج
متلازمة برادر ويلي ليس له علاج، ولكن العديد من العلاجات المتبعة لتخفيف أعراض الحالة.ففي مرحلة الطفولة، يجب أن يخضع المصابين للعلاج لتحسين قوة العضلات. و أيضا التدريب والمساعدة على النطق.[12] خلال سنوات الدراسة، يستفيد الأطفال من المدارس عالية التنظيم فضلا عن المساعدة الخارجية. السمنة المفرطة هي أكبر المشاكل المصاحبة للمتلازمة .[6] حقن هرمون النمو المأشوب يوميا حددت للأطفال مع المتلازمة. هرمون النمو يدعم النمو الخطي ويزيد من كتلة العضلات، وقد يقلل من الانهماك في الأكل وازدياد الوزن.[31][32][33] بسب السمنة المفرطة، انقطاع النفس النومي من العقابيل الشائعة، و غالبا ما يلزم آلة ضغط المجرى الهوائي الإيجابي. وقد يأتي وقت يحتاج فيه الأشخاص الذين تم تشخيصهم بالمتلازمة إلى عمليات جراحية. تم إثبات أن عملية المجازة المعدية غير ناجحة في علاج السمنة. المرضى المصابين بالمتلازمة يملكون قدرة عالية على تحمل الألم؛ وبهذا من الممكن أن يعانوا من أعراض هامة في البطن مثل التهاب معدة حاد، التهاب الزائدة أو التهاب المرارة ولا يكون على علم به الا لاحقا.
المجتمع والثقافة
%252C_de_Juan_Carre%C3%B1o_de_Miranda..jpg.webp)
على الرغم من ندرتها، إلى أنه تم الإشارة لمتلازمة برادر-ويلي في الثقافة الشعبية، جزئيا بسبب لفت الانتباه للسمنة والشهية النهمة وهما من إحدى الأعراض. لقد تم تصوير وتوثيق متلازمة برادر-ويلي مرات عدة على التلفاز. تم عرض شخصية خيالية تملك المتلازمة في حلقة "كلب يأكل كلب"Dog Eat Dog- من المسلسل التلفزيوني CSI: Crime Scene Investigation تم بثها في 24 نوفمبر عام 2005.[35] قامت وسائل اعلام المملكة المتحدة في يوليو 2007 على القناة الرابعة ,Channel 4وثائقي سنة 2006 Can't Stop Eating, الذي يحيط حياة شخصين يملكان متلازمة برادر-ويلي، جوي و تمارا.[36] في 9 مايو عام 2010 حلقة من برنامج Extreme Makeover: Home Edition,Sheryl Crow ساعد Ty Penningtonفي إعادة بناء بيت لعائلة يعاني أصغر أفرادها من متلازمة برادر-ويلي.[37] في 22 مارس عام 2012 حلقة من برنامج Mystery Diagnosisبالعربية التشخيص الغامض الذي يعرض على قناة ديسكافري الصحة،ConorHeybachالذي يملك المتلازمة يشارك قصة تشخيصه للمتلازمة.[38] في ديسمبر من العام 2011، Taipei Timesفي تايوانسلطت الضوء على مأساة سائق التاكسي الذي قتل نفسه وقتل ابنه ذي التسع سنوات والذي يملك المتلازمة، وقد وصفت الشرطة الحادثة ب "قتل-انتحار محتمل".[39] الممثلة ميايم بياليك- النجمة السابقة لمسلسل الزهرة Blossom وحاليا تلعب دور عالمة أعصاب تسمى Amy Farrah Fowler في The Big Bang Theory بالعربية نظرية الانفجار العظيم- أكملت شهادة الدكتوراة عام 2007 باطروحة عن متلازمة برادر-ويلي:" التنظيم الوطائي وعلاقته مع سلوكيات maladaptive، الاضطراب الوسواسي القهري، , affiliative والشبع في متلازمة برادر-ويلي."
انظر ايضا
المراجع
- "Prader-Labhardt-Willi syndrome". Whonamedit?. مؤرشف من الأصل في 27 أبريل 2019. اطلع عليه بتاريخ 20 أغسطس 2016. الوسيط
|CitationClass=تم تجاهله (مساعدة) - https://www.ncbi.nlm.nih.gov/books/NBK1330/
- "Prader-Willi syndrome". Genetics Home Reference. June 2014. مؤرشف من الأصل في 28 أبريل 2019. اطلع عليه بتاريخ 19 أغسطس 2016. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Prader-Willi Syndrome (PWS): Other FAQs". NICHD. 01/14/2014. مؤرشف من الأصل في 22 ديسمبر 2017. اطلع عليه بتاريخ 19 أغسطس 2016. الوسيط
|CitationClass=تم تجاهله (مساعدة); تحقق من التاريخ في:|تاريخ=(مساعدة) - "Angelman syndrome". Genetic Home Reference. May 2015. مؤرشف من الأصل في 17 مايو 2019. اطلع عليه بتاريخ 20 أغسطس 2016. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Cassidy, S. B., & Driscoll, D. J. (2009). Prader–Willi syndrome. European Journal of Human Genetics, 17(1), 3–13. http://doi.org/10.1038/ejhg.2008.165
- Curfs LM, Fryns JP (1992). "Prader-Willi syndrome: a review with special attention to the cognitive and behavioral profile". Birth Defects Orig. Artic. Ser. 28 (1): 99–104. PMID 1340242. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Cassidy SB (1997). "Prader-Willi syndrome". Journal of Medical Genetics. 34 (11): 917–23. doi:10.1136/jmg.34.11.917. PMC 1051120. PMID 9391886. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Udwin O (November 1998). "Prader-Willi syndrome: Psychological and behavioural characteristics". Contact a Family. مؤرشف من الأصل في 8 مارس 2012. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Holm VA, Cassidy SB, Butler MG, Hanchett JM, Greenswag LR, Whitman BY, Greenberg F (1993). "Prader-Willi syndrome: consensus diagnostic criteria". Pediatrics. 91 (2): 398–402. PMID 8424017. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Whittington J, Holland A, Webb T, Butler J, Clarke D, Boer H (February 2004). "Cognitive abilities and genotype in a population-based sample of people with Prader-Willi syndrome". J Intellect Disabil Res. 48 (Pt 2): 172–87. doi:10.1111/j.1365-2788.2004.00556.x. PMID 14723659. الوسيط
|CitationClass=تم تجاهله (مساعدة) - What are the treatments for Prader-Willi syndrome (PWS)? نسخة محفوظة 22 ديسمبر 2017 على موقع واي باك مشين.
- Nordqvist, Christian (March 15, 2010). "What Is Prader-Willi Syndrome? What Causes Prader-Willi Syndrome?". Medical News Today. MediLexicon International. مؤرشف من الأصل في 13 أبريل 2019. اطلع عليه بتاريخ December 4, 2012. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Cummings, D.E., Purnell, J.Q., Vaisse, C., Foster, K.E., Frayo, R.S., Schwartz, M.W., Basdevant, A., & Weigle, D.S. (2002). "Elevated plasma ghrelin levels in Prader Willi syndrome". Nature Medicine. 8: 643–644. doi:10.1038/nm0702-643. الوسيط
|CitationClass=تم تجاهله (مساعدة)صيانة CS1: أسماء متعددة: قائمة المؤلفون (link) - Clark DJ, Boer H, Webb T (1995). "General and behavioural aspects of PWS: a review". Mental Health Research. 8 (195): 38–49. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Cassidy SB, Devi A, Mukaida C (1994). "Aging in PWS: 232 patients over age 30 years". Proc. Greenwood Genetic Centre. 13: 102–3. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Hered RW, Rogers S, Zang YF, Biglan AW (1988). "Ophthalmologic features of Prader-Willi syndrome". J Pediatr Ophthalmol Strabismus. 25 (3): 145–50. PMID 3397859. الوسيط
|CitationClass=تم تجاهله (مساعدة) - الوراثة المندلية البشرية عبر الإنترنت (OMIM) Prader-Willi Syndrome; PWS -17627
- de los Santos T, Schweizer J, Rees CA, Francke U (November 2000). "Small evolutionarily conserved RNA, resembling C/D box small nucleolar RNA, is transcribed from PWCR1, a novel imprinted gene in the Prader-Willi deletion region, which Is highly expressed in brain". American Journal of Human Genetics. 67 (5): 1067–82. doi:10.1086/303106. PMC 1288549. PMID 11007541. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Cavaillé J, Buiting K, Kiefmann M, Lalande M, Brannan CI, Horsthemke B, Bachellerie JP, Brosius J, Hüttenhofer A (December 2000). "Identification of brain-specific and imprinted small nucleolar RNA genes exhibiting an unusual genomic organization". Proc. Natl. Acad. Sci. USA. 97 (26): 14311–6. doi:10.1073/pnas.250426397. PMC 18915. PMID 11106375. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Buiting, K; Saitoh, S; Gross, S; Dittrich, B; Schwartz, S; Nicholls, RD; Horsthemke, B (April 1995). "Inherited microdeletions in the Angelman and Prader-Willi syndromes define an imprinting centre on human chromosome 15". Nature Genetics. 9 (4): 395–400. doi:10.1038/ng0495-395. PMID 7795645. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Major breakthrough in understanding Prader-Willi syndrome, a parental imprinting disorder". Medicalxpress.com. مؤرشف من الأصل في 2 أبريل 2019. اطلع عليه بتاريخ 18 يونيو 2015. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Runte M, Varon R, Horn D, Horsthemke B, Buiting K (2005). "Exclusion of the C/D box snoRNA gene cluster HBII-52 from a major role in Prader-Willi syndrome". Hum Genet. 116 (3): 228–30. doi:10.1007/s00439-004-1219-2. PMID 15565282. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Skryabin BV, Gubar LV, Seeger B, Pfeiffer J, Handel S, Robeck T, Karpova E, Rozhdestvensky TS, Brosius J (2007). "Deletion of the MBII-85 snoRNA gene cluster in mice results in postnatal growth retardation". PLoS Genet. 3 (12): e235. doi:10.1371/journal.pgen.0030235. PMC 2323313. PMID 18166085. مؤرشف من الأصل في 5 مارس 2014. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Sahoo T, del Gaudio D, German JR, Shinawi M, Peters SU, Person RE, Garnica A, Cheung SW, Beaudet AL (2008). "Prader-Willi phenotype caused by paternal deficiency for the HBII-85 C/D box small nucleolar RNA cluster". Nat Genet. 40 (6): 719–21. doi:10.1038/ng.158. PMC 2705197. PMID 18500341. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Ding F, Li HH, Zhang S, Solomon NM, Camper SA, Cohen P, Francke U (2008). Akbarian, Schahram (المحرر). "SnoRNA Snord116 (Pwcr1/MBII-85) deletion causes growth deficiency and hyperphagia in mice". PLoS ONE. 3 (3): e1709. doi:10.1371/journal.pone.0001709. PMC 2248623. PMID 18320030. الوسيط
|CitationClass=تم تجاهله (مساعدة)صيانة CS1: أسماء متعددة: قائمة المؤلفون (link) - Ding F, Prints Y, Dhar MS, Johnson DK, Garnacho-Montero C, Nicholls RD, Francke U (2005). "Lack of Pwcr1/MBII-85 snoRNA is critical for neonatal lethality in Prader-Willi syndrome mouse models". Mamm Genome. 16 (6): 424–31. doi:10.1007/s00335-005-2460-2. PMID 16075369. الوسيط
|CitationClass=تم تجاهله (مساعدة) - de Smith AJ, Purmann C, Walters RG, Ellis RJ, Holder SE, Van Haelst MM, Brady AF, Fairbrother UL, Dattani M, Keogh JM, Henning E, Yeo GS, O'Rahilly S, Froguel P, Farooqi IS, Blakemore AI (June 2009). "A Deletion of the HBII-85 Class of Small Nucleolar RNAs (snoRNAs) is Associated with Hyperphagia, Obesity and Hypogonadism". Hum. Mol. Genet. 18 (17): 3257–65. doi:10.1093/hmg/ddp263. PMC 2722987. PMID 19498035. مؤرشف من الأصل في 26 أبريل 2020. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Killeen, Anthony A. (2004). "Genetic Inheritance". Principles of Molecular Pathology. Humana Press. صفحة 41. ISBN 978-1-58829-085-4. مؤرشف من الأصل في 29 يونيو 2014. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Tweed, Katherine (September 2009). "Shawn Cooper Struggles with Prader Willi Syndrome". AOL Health. مؤرشف من الأصل في 9 سبتمبر 2009. اطلع عليه بتاريخ September 2009. الوسيط
|CitationClass=تم تجاهله (مساعدة); تحقق من التاريخ في:|تاريخ الوصول=(مساعدة) - Davies PS, Evans S, Broomhead S, Clough H, Day JM, Laidlaw A, Barnes ND (May 1998). "Effect of growth hormone on height, weight, and body composition in Prader-Willi syndrome". Arch. Dis. Child. 78 (5): 474–6. doi:10.1136/adc.78.5.474. PMC 1717576. PMID 9659098. مؤرشف من الأصل في 11 مايو 2020. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Carrel AL, Myers SE, Whitman BY, Allen DB (April 2002). "Benefits of long-term GH therapy in Prader-Willi syndrome: a 4-year study". J. Clin. Endocrinol. Metab. 87 (4): 1581–5. doi:10.1210/jc.87.4.1581. PMID 11932286. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Höybye C, Hilding A, Jacobsson H, Thorén M (May 2003). "Growth hormone treatment improves body composition in adults with Prader-Willi syndrome". Clin. Endocrinol. (Oxf). 58 (5): 653–61. doi:10.1046/j.1365-2265.2003.01769.x. PMID 12699450. مؤرشف من الأصل في 26 أبريل 2020. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Mary Jones. "Case Study: Cataplexy and SOREMPs Without Excessive Daytime Sleepiness in Prader Willi Syndrome. Is This the Beginning of Narcolepsy in a Five Year Old?". European Society of Sleep Technologists. مؤرشف من الأصل في 6 يونيو 2017. اطلع عليه بتاريخ April 6, 2009. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Dog Eat Dog". Csifiles.com. مؤرشف من الأصل في 15 ديسمبر 2018. اطلع عليه بتاريخ 12 يونيو 2009. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Can't Stop Eating". Channel4.com. 2006. مؤرشف من الأصل في 29 مارس 2010. اطلع عليه بتاريخ 12 يونيو 2009. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Extreme Makeover: Home Edition Articles on AOL TV". Aoltv.com. مؤرشف من الأصل في 14 ديسمبر 2018. اطلع عليه بتاريخ 18 يونيو 2015. الوسيط
|CitationClass=تم تجاهله (مساعدة) - تم أرشفته يوليو 14, 2014 بواسطة آلة واي باك نسخة محفوظة 14 يوليو 2014 على موقع واي باك مشين. [وصلة مكسورة]
- Group urges more support for Prader-Willi sufferers, Taipei Times. Published December 24, 2011. Retrieved May 27, 2012. نسخة محفوظة 04 يونيو 2017 على موقع واي باك مشين.
Cassidy, S. B., & Driscoll, D. J. (2009). Prader–Willi syndrome. European Journal of Human Genetics, 17(1), 3–13. http://doi.org/10.1038/ejhg.2008.165
 بوابة طب
بوابة طب
