متلازمة أشر-هولجرين
متلازمة أشر-هولجرين أو متلازمة أشر أو متلازمة هولجرين أو متلازمة التهاب الشبكية الصباغي وخلل السمع أو متلازمة حثل الشبكية وخلل السمع (بالإنجليزية: Usher-Hallgren syndrome) هي مرض وراثي نادر ينشأ عن تحورات في حوالي 11 جين، ويعاني بسببها المريض من فقد للسمع والإبصار، ولا يوجد لها علاج شافي حتى الآن.
| متلازمة أشر-هولجرين | |
|---|---|
| معلومات عامة | |
| الاختصاص | طب العيون |
| من أنواع | اضطراب صبغي جسدي متنحي ، ومتلازمة |
ميكانيكية المرض
تحتوي شبكية العين على نوعين من الخلايا هما العصي (rods) ووظيفتها الرئيسية هي دعم الرؤية في الضوء الخافت ورؤية الأبيض والأسود، والمخاريط (cones) ووظيفتها دعم الرؤية بالنهار وإدراك الألوان. في متلازمة أشر-هولجرين تصاب الشبكية بما يعرف بالتهاب الشبكية الصباغي حيث تفقد خلايا العصي وظيفتها تدريجيًا فيعاني المريض من صعوبة أو استحالة الرؤية في الضوء الخافت. ومع تقدم الحالة المرضية تتأثر خلايا المخاريط كذلك، فيصاب النظر بحالة شديدة من الضعف بمرور من 30 إلى 40 سنة. أما ضعف أو فقد السمع فيحدث نتيجة خلل تخليق وتكوين الأذن الداخلية.
تاريخيًا
قام الطبيب الألماني ألبريشت فون جريف مؤسس طب العيون في ألمانيا بوصف حالة مرضية يصاب فيها المريض بفقد السمع و التهاب الشبكية الصباغي عام 1858, وقد سجّل وجود أخوين مصابين بنفس الحالة مما يرجح كونها مرضًا وراثيَا.[1] ولاحقًا سميت متلازمة أشر-هولجرين نسبة إلى الطبيب الاسكتلاندي تشارليز أشر أخصائي طب العيون, والذي قام ببحث سبب المتلازمة وطريقة وراثتها بدراسة حالة 69 مريض.[2]
وراثة المرض
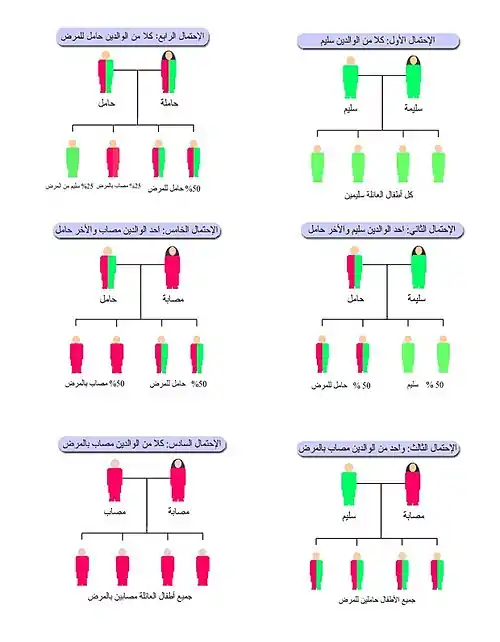
تنتقل متلازمة أشر-هولجرين بشكل صفة متنحية، بمعنى أن الشخص الحامل لجين متلازمة أشر-هولجرين ليس مصابًا بالمرض ولا تظهر عليه أي أعراض ولن يظهر عليه فيما بعد، ولكن هناك احتمالية لنقل هذا الجين لأطفاله في المستقبل عن طريق الاحتمالات التالية:
1-إن زاوج شخص يحمل صفة المتلازمة من شخص سليم فإن احتمالات الإنجاب تكون:
- 50% أطفال سليمين.
- 50% أطفال يحملون صفة المتلازمة
وبذلك فليس هناك خطر من إنجاب طفل مريض بمتلازمة أشر-هولجرين
2-إن زواج شخص يحمل صفة متلازمة أشر-هولجرين من شخص يحمل أيضا الصفة فإن احتمالات الإنجاب تكون:
- 25% أطفال سليمين.
- 50% أطفال يحملون الصفة.
- 25% أطفال مرضى بمتلازمة أشر-هولجرين.
3-إن زواج شخص مريض بمتلازمة أشر-هولجرين من شخص سليم فإن احتمالات الإنجاب تكون:
- 100% أطفال يحملون الصفة.
وبذلك فليس هناك خطر من إنجاب طفل مريض بالمتلازمة.
4-إن زواج شخص مريض بمتلازمة أشر-هولجرين من شخص يحمل الصفة فإن احتمالات الإنجاب تكون:
- 50% أطفال يحملون الصفة.
- 50% أطفال مرضى بالمتلازمة.
وقد ثبت وجود 13 جين مختلف مسئول عن انتقال متلازمة أشر-هولجرين.وتنصح العائلات التي تحتوي على أفراد مصابين بمتلازمة أشر-هولجرين بإجراء فحوصات قبل التزاوج لتجنب انتقال المرض إلى الأبناء.[3]
معدلات الحدوث
تصيب متلازمة أشر-هولجرين واحدًا من بين 23.000 شخص في الولايات المتحدة الأمريكية,[4] وواحدًا من بين 12.500 شخص في ألمانيا.[5] وتمثل متلازمة أشر-هولجرين 1\6 حالات التهاب الشبكية الصباغي.
الأعراض والعلامات
تصنّف متلازمة أشر-هولجرين إلى 3 أنواع حسب اختلاف الأعراض والجينات المسؤولة عن انتقال المرض وليس حسب شدته بتاتًا:
النوع الأول
يولد الأطفال المصابون بمتلازمة أشر-هولجرين فاقدين للسمع تمامًا، كما يعانون من اختلالات في تخليق الجزء الخاص من الجهاز السمعي المسؤول عن عملية الاتزان والمسمى بـ(الجهاز الدهليزي) مما يعرضهم في الطفولة لبطء التطور الحركي كتعلم المشي. بالإضافة إلى إصابتهم بالتهاب الشبكية الصباغي والذي يؤثر سلبًا على الرؤية في الضوء الخافت وليلًا.
ينتشر النوع الأول من متلازمة أشر-هولجرين بين شعوب يهود الأشكناز الذين يعيشون في وسط وشرق أوروبا وشعوب الأكاديين وهم يسكنون مناطق من فرنسا.
النوع الثاني
يعاني مرضى النوع الثاني من متلازمة أشر-هولجرين من ضعف وصعوبة السمع وليس فقد السمع التام، كما أنهم لا يعانون من أي خلل في الجهاز الدهليزي, ويعتقد بأن النوع الثاني أكثر انتشارًا من النوع الأول وعدد حالاته تصل إلى 3 أضعاف أعداد حالات النوع الأول ولكن بسبب صعوبة تشخيصه تظهر أعداد حالات النوع الثاني كأنها مساوية لأعداد حالات النوع الأول.
النوع الثالث
يعاني مرضى النوع الثاني من متلازمة أشر-هولجرين من ضعف شديد جدًا بالسمع ويعاني نصف الحالات من اختلال الجهاز الدهليزي. وينتشر النوع الثالث في فنلندا.
العلاج
لا يوجد علاج شافي حتى الآن لمتلازمة أشر-هولجرين وتعقد الكثير من الآمال على العلاج الجيني الذي لازال تحت البحث.
مصادر
- von Gräfe A (1858). "Exceptionelles Verhalten des Gesichtsfeldes bei Pigmententartung der Netzhaut". Archiv für Ophthalmologie. 4: 250–253. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Usher C (1914). "On the inheritance of Retinitis pigmentosa with notes of cases". Roy. Lond. Ophthalmol. Hosp. Rep. 19: 130–236. الوسيط
|CitationClass=تم تجاهله (مساعدة) - How Can Fanconi Anemia Be Prevented? - NHLBI, NIH نسخة محفوظة 28 يوليو 2017 على موقع واي باك مشين.
- Boughman J, Vernon M, Shaver K (1983). "Usher syndrome: Definition and estimate of prevalence from two high-risk populations". Journal of Chronic Diseases. 36 (8): 595–603. doi:10.1016/0021-9681(83)90147-9. PMID 6885960. الوسيط
|CitationClass=تم تجاهله (مساعدة)صيانة CS1: أسماء متعددة: قائمة المؤلفون (link) - Otterstedde CR, Spandau U, Blankenagel A, Kimberling WJ, Reisser C (2001). "A new clinical classication for Usher's syndrome based on a new subtype of Usher's syndrome type I". Laryngoscope. 111 (1): 84–86. doi:10.1097/00005537-200101000-00014. PMID 11192904. الوسيط
|CitationClass=تم تجاهله (مساعدة)صيانة CS1: أسماء متعددة: قائمة المؤلفون (link)
 بوابة طب
بوابة طب