كثرة الكريات الإهليلجية الوراثية
كثرة الكريات الإهليلجية الوراثية، تُدعَى أيضًا كثرة الكريات البيضوية الوراثية، هي اضطراب دموي موروث يكون فيه عدد كبير غير طبيعي من كريات الدم الحمراء لدى الشخص ذات شكل إهليلجي بدلًا من الشكل القرصي مقعر الوجهين. يُشار أحيانًا إلى هذه الكريات الحمراء المميزة شكليًا بالكريات الإهليلجية أو الكريات البيضوية. إنها إحدى عيوب عديدة في غشاء الكرية الحمراء. في شكله الشديد، يؤهب هذا الاضطراب لحدوث فقر الدم. رغم أنه يُعتبَر حالة مرضية لدى البشر، إلا أن كثرة الكريات الإهليلجية تكون طبيعية لدى الجمليات.
| كثرة الكريات الإهليلجية الوراثية | |
|---|---|
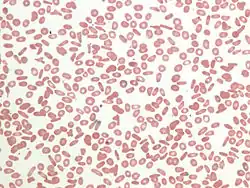 شريحة دم تظهر عددًا كبيرًا من كثرة الكريات الإهليلجية شريحة دم تظهر عددًا كبيرًا من كثرة الكريات الإهليلجية | |
| معلومات عامة | |
| الاختصاص | علم الدم |
| من أنواع | مرض دموي |
| المظهر السريري | |
| الأعراض | كرية إهليلجية |
الفيزيولوجيا
كثرة الكريات الإهليلجية الشائع
رُبِط عدد من المورثات بحدوث كثرة الكريات الإهليلجية الوراثي (العديد من الباحثون يتهمون نفس المورثة المسؤولة عن كثرة الكريات المكوّرة الوراثية).
لهذه الطفرات نتيجة نهائية مشتركة، إنها تزعزع استقرار سقالات الهيكل الخلوي للخلية. هذا الاستقرار مهم بشكل خاص للكريات الحمراء لأنها تتعرض باستمرار لتأثير قوى قص مُشوِّهة. تعبر الكريات الحمراء الشعيرات الدموية –التي قد يكون عرضها 2 – 3 ميكرون- بوصفها قرصًا مقعر الوجهين، فتُجبَر على اتخاذ الشكل الإهليلجي كي تتمكن من العبور. في الحالة الطبيعية، يقتصر حدوث هذا التشوه على الفترة التي تكون خلالها الخلية ضمن الشعيرة الدموية، لكن في حالة الإصابة بكثرة الكريات الإهليلجية الوراثية سيؤدي عدم استقرار الهيكل الخلوي هذا إلى تشوه الكريات الحمراء أثناء عبورها الشعيرة وستبقى إهيليجية الشكل إلى الأبد. يبتلع الطحال الكريات الإهليلجية ويزيلها من الدوران الدموي بعمر أصغر من الذي يحدث فيه الابتلاع في الحالة الطبيعية ما يعني أن عمر الكريات الحمراء لدى المصابين بكثرة الكريات الإهليلجية الوراثية يكون أقصر من المتوسط (العمر المتوسط للكريات الحمراء لدى الشخص الطبيعي 120 يوم أو أكثر).
- EL2، و EL3: أشيع عيبين مورثيين (يوجدان في ثلثي حالات كثرة الكريات الإهليلجية الوراثية) يحدثان في مورثات عديدي الببتيد ألفا-سبيكترين أو بيتا-سبيكترين. يجتمع هذين الببتيدين مع بضعهما البعض في الجسم الحي لتشكيل ثنائيات متغايرة ألفا-بيتا. تجتمع هذه الثنائيات المتغايرة ألفا-بيتا لتشكيل رباعيات سبيكترين. إن رباعيات السبيكترين هذه من بين تحت الوحدات الهيكلية الأساسية للهيكل الخلوي في جميع خلايا الجسم. رغم وجود الكثير من التنوعات بين الأفراد، من الصحيح بشكل عام أن طفرات ألفا-سبيكترين تؤدي إلى عدم القدرة ألفا-سبيكترين على التفاعل بشكل صحيح مع بيتا-سبيكترين لتشكيل الثنائيات المتغايرة. بالمقابل، من الصحيح بشكل عام أن طفرات بيتا-سبيكترين تؤدي إلى ثنائيات متغاير ألفا-بيتا تكون غير قادرة على التجمع لتشكيل رباعيات السبيكترين.[1] في كلتا الحالتين، تكون النتيجة النهائية ضعفًا في الهيكل الخلوي للخلية. عادة ما يكون الأشخاص المصابين بطفرة واحدة في أحد مورثات السبيكترين غير عرضيين، لكن متماثلي اللواقح (مثماثلي الزيجوت) أو متخالفي اللواقح (على سبيل المثال، متخالفي اللواقح بالنسبة لطفرتين مسببتين لكثرة الكريات الإهليلجية الوراثية) يكون لديهم عدم استقرار غشاء خلوي كافٍ حتى يظهر لديهم فقر دم مهم سريريًا.
- EL1: طفرات الشريط 4.1 أقل شيوعًا من طفرات السبيكترين. يجب أن ترتبط رباعيات السبيكترين مع الأكتين لإنشاء سقالات هيكل خلوي مناسبة، والشريط 4.1 هو بروتين هام يشارك في استقرار الرابط بين السبيكترين والأكتين. بشكل مشابه لطفرات السبيكترين، تسبب طفرات الشريط 4.1 فقر دم خفيف في حالة تخالف اللواقح، وفقر دم شديد في حالة تماثل اللواقح.
- EL4: ترتبط كثرة الكريات البيضوية لجنوب شرق آسيا مع بروتين الشريط 3.
- مجموعة أخرى من الطفرات التي تؤدي إلى حدوث كثرة الكريات الإهليلجية هي تلك التي تسبب أعواز الغليكوفورين C. هناك ثلاثة أنماط ظاهرية تنتج عن الغليكوفورين C غير الطبيعي، تدعى هذه الأنماط: جيربيش، ويوس، وليتش. فقط الأندر من بين هذه الأنماط الثلاثة، والنمط الظاهري لليتش، يسبب حدوث كثرة الكريات الإهليلجية. للغليكوفورين C وظيفة تثبيت الشريط 4.1 إلى غشاء الخلية. يُعتقَد أن كثرة الكريات الإهليلجية في عوز الغليكوفورين C هي بالحقيقة نتيجة عيب في الشريط 4.1، لأن الأشخاص الذين لديهم عوز الغليكوفورين C يكون لديهم أيضًا نقص في الشريط 4.1 داخل الخلايا (ربما بسبب تقص عدد المواقع الرابطة للشريط 4.1 في غياب الغليكوفورين C)
تميل وراثة طفرات متعددة للتداخل مع أمراض أخطر. على سبيل المثال، يحدث أشيع نمط ظاهري مسؤول عن كثرة الكريات الحمراء الوراثية عندما يرث الشخص المصاب طفرة ألفا-سبيكترين من أحد الوالدين (على سبيل المثال، يكون أحد الوالدين مصاب بكثرة الكريات الإهليلجية الوراثية) وينقل الوالد الآخر عيبًا لم يُحدَّد بعد يدفع خلايا الشخص المصاب لإنتاج ألفا-سبيكترين معيب بدلًا من ألفا-سبيكترين طبيعي.
التشخيص
يوضع تشخيص كثرة الكريات الإهليلجية الوراثية بالربط بين تاريخ المرض في العائلة مع التظاهر السريري المناسب، ويكون تأكيد التشخيص بشريحة الدم. يتطلب التشخيص بصورة عامة أن تكون على الأقل 25% من الكريات الحمراء في العينة إهليلجية الشكل، رغم أن النسبة المُشاهَدة من الكريات الإهليلجية قد تصل إلى 100%. هذا بعكس باقي الناس الذين يشيع أن يكون لديهم ما يصل إلى 15% من الكريات الحمراء إهليلجية الشكل.[2]
إن بقي بعض الشك فيما يتعلق بالتشخيص، قد يتضمن التشخيص النهائي اختبار الهشاشية التناضحية، وهو اختبار انحلال دم ذاتي، وفحص البروتين المباشر بواسطة الفصل الكهربائي الهلامي.[3]
المراجع
- McMullin MF (1999). "The molecular basis of disorders of the red cell membrane". J. Clin. Pathol. 52 (4): 245–8. doi:10.1136/jcp.52.4.245. PMC 501324. PMID 10474512. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Gerard M. Doherty (2010). Current Diagnosis & Treatment - Surgery (الطبعة 13th). McGraw Hill Professional. صفحات 204–5. ISBN 978-0-07-163515-8. مؤرشف من الأصل في 18 أبريل 2020. اطلع عليه بتاريخ 05 مايو 2011. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Robert S. Hillman; Kenneth A. Ault; Henry M. Rinder (2005). Hematology in clinical practice: a guide to diagnosis and management (الطبعة 4th). McGraw-Hill Professional. صفحة 147. ISBN 978-0-07-144035-6. مؤرشف من الأصل في 18 أبريل 2020. اطلع عليه بتاريخ 05 مايو 2011. الوسيط
|CitationClass=تم تجاهله (مساعدة)
 صور وملفات صوتية من كومنز
صور وملفات صوتية من كومنز
 بوابة طب
بوابة طب