علم الجينوم الوظيفي
علم الجينوم الوظيفي هو علم يبحث في مجال البيولوجيا الجزيئية الذي يحاول وصف وظائف الجينات والبروتينات الوظائف والتفاعلات. تستفيد الجينوم الوظيفية من البيانات الهائلة الناتجة عن المشروعات الجينومية والنسخة النصية (مثل مشاريع تسلسل الجينوم وتسلسل الحمض النووي ). تركز الجينوم الوظيفية على الجوانب الديناميكية مثل نسخ الجينات والترجمة وتنظيم التعبير الجيني وتفاعلات البروتين - البروتين ، على عكس الجوانب الثابتة للمعلومات الجينية مثل تسلسل الحمض النووي أوالبنية. من الخصائص الرئيسية لدراسات الجينوم الوظيفية النطاق الواسع للجينوم تجاه هذه الأسئلة، والذي يشتمل عمومًا على طرق إنتاجية عالية بدلاً من طريقة "جين بواسطة جين" .

تعريف وأهداف علم الجينوم الوظيفي
من أجل فهم علم الجينوم الوظيفي، من المهم تحديد الوظيفة أولاً. في ورقتهم [1] Graur et al. تحديد الوظيفة بطريقتين ممكنتين. هذه هي "التأثير المحدد" و "الدور السببي". تشير وظيفة "Selected Effect" إلى الوظيفة التي يتم تحديد سمة لها (DNA ، RNA ، بروتين إلخ). تشير وظيفة "الدور السببي" إلى الوظيفة التي تعد سمة كافية وضرورية لها. عادة ما تختبر الجينوم الوظيفي تعريف "الدور السببي" للوظيفة.
الهدف من علم الجينوم الوظيفي هو فهم وظيفة الجينات أو البروتينات، وفي النهاية كل مكونات الجينوم. غالبًا ما يستخدم مصطلح الجينوم الوظيفي للإشارة إلى العديد من الأساليب الفنية لدراسة جينات الكائن الحي والبروتينات ، بما في ذلك "الخواص الكيميائية الحيوية و / أو الخلوية و / أو الفسيولوجية لكل منتج جين" [2] حين أن بعض المؤلفين يشملون دراسة العناصر اللاجينية في تعريفهم.[3] قد تشمل الجينوم الوظيفية أيضًا دراسات عن التباين الوراثي الطبيعي مع مرور الوقت (مثل تطور الكائن الحي) أو الفضاء (مثل مناطق متعلقة به) ، وكذلك الاضطرابات الوظيفية مثل الطفرات.
الهدف من علم الجينوم الوظيفي هو توليد وتوليف المعرفة الجينومية والبروتينية في فهم الخصائص الديناميكية للكائن الحي. يمكن أن يوفر هذا صورة أكثر اكتمالا عن كيفية تحديد الجينوم الوظيفية مقارنة بدراسات الجينات المنفردة. غالبًا ما يكون تكامل بيانات الجينوم الوظيفية جزءًا من مناهج بيولوجيا الأنظمة .
التقنيات والتطبيقات
تشتمل الجينوم الوظيفية على الجوانب المتعلقة بوظيفة الجينوم نفسه، مثل الطفرة وتعدد الأشكال (مثل تحليل تعدد الأشكال النوكليوتيد الفردي (SNP)) ، وكذلك قياس الأنشطة الجزيئية. يشتمل الأخير على عدد من " omics " مثل transcriptomics ( التعبير الجيني ) ، والبروتينات (إنتاج البروتينات والأيض . تستخدم الجينوم الوظيفية في الغالب تقنيات متعددة لقياس مدى وفرة العديد من أو كل منتجات الجينات مثل mRNAs أو البروتينات ضمن عينة بيولوجية . قد يختبر نهج الجينوم الوظيفي الأكثر تركيزًا وظيفة جميع المتغيرات في جين واحد وتحديد تأثيرات المسوخ باستخدام التسلسل كقراءة للنشاط. وتسعى طرق القياس هذه معًا لتقدير العمليات البيولوجية المختلفة وتحسين فهمنا لوظائف الجينات والبروتين والتفاعلات.
خريطة التفاعل الوراثي
يمكن استخدام الحذف المنهجي للجينات أو تثبيط التعبير الجيني لتحديد الجينات ذات الصلة، حتى لو لم تتفاعل بشكل فيزيائي. يشير Epistasis إلى حقيقة أن التأثيرات لاثنين من طرق خروج الجينات المختلفة قد لا تكون مضافة ؛ أي أن النمط الظاهري الذي ينتج عند تثبيط جينين قد يكون مختلفًا عن مجموع تأثيرات الضربة القاضية المفردة.
تفاعلات الحمض النووي / البروتين
تلعب البروتينات مثل عوامل النسخ دورًا رئيسيًا في تنظيم التعبير الجيني. لفهم كيفية تنظيم التعبير الجيني، من الضروري تحديد تسلسل الحمض النووي مع الذي يتفاعل معه . تم تطوير تقنيات لتحديد مواقع تفاعلات بروتين الحمض النووي. وتشمل هذه التسلسل رقاقة، CUT & RUN التسلسل وبطاقات الاتصال.
فحوصات وصول الحمض النووي
تم تطوير مقاييس لتحديد مناطق الجينوم التي يمكن الوصول إليها. هذه المناطق من الكروماتين المفتوحة هي المناطق التنظيمية المرشحة. تشمل هذه المقايسات ATAC-seq و DNase-Seq و FAIRE-Seq .
المصفوفات الدقيقة
تقوم المصفوفات الدقيقة بقياس كمية mRNA في عينة تتوافق مع جين معين أو تسلسل الحمض النووي للجين. يتم تجميد تسلسلات المجس على سطح صلب ويسمح لها بالتهجين باستخدام mRNA "الهدف" المسمى بالفلورسنت. تتناسب شدة الموضعية في البقعة مع مقدار التتابع المستهدف الذي تم تهجينه لتلك البقعة، وبالتالي إلى وفرة تسلسل mRNA في العينة. تسمح المصفوفات المجهرية بتحديد الجينات المرشحة المشاركة في عملية معينة بناءً على التباين بين مستويات النسخ للظروف المختلفة وأنماط التعبير المشتركة مع جينات الوظيفة المعروفة.
التحليل التسلسلي للتعبير الجيني (SAGE) هو طريقة بديلة للتحليل تعتمد على تسلسل الحمض النووي الريبي (RNA) بدلاً من التهجين. يعتمد SAGE على تسلسل 10-17 علامات ازواج أساسية وهي فريدة لكل جين. يتم إنتاج هذه العلامات من poly-A mRNA ويتم ربطها من طرف إلى طرف قبل التسلسل. يعطي SAGE قياسًا غير متحيز لعدد النصوص لكل خلية، لأنه لا يعتمد على معرفة مسبقة بما تدرسه النصوص (كما تفعل المصفوفات المجهرية).
تسلسل الحمض النووي
استحوذ تسلسل الحمض النووي على تقنيات المصفوفات الدقيقة و SAGE في السنوات الأخيرة، كما لوحظ في عام 2016 ، وأصبح الطريقة الأكثر فعالية لدراسة النسخ والتعبير الجيني. ويتم ذلك عادة من خلال تسلسل الجيل التالي .[4]
مجموعة فرعية من RNA's المتسلسلة عبارة عن RNA's صغيرة، وهي فئة من جزيئات RNA غير المشفرة والتي تعد من الجهات المنظمة الرئيسية لشل الجينات النصية وما بعد النسخية أوشل RNA . يمثل تسلسل الجيل التالي الأداة المعيارية الذهبية لاكتشاف تحليل الحمض النووي الريبي (RNA) غير المشفر وتحليله وتعبيره.
فحوصات مراسل موازية على نطاق واسع (MPRAs)
فحوصات المراسل الموازية على نطاق واسع هي تقنية لاختبار النشاط المنظم ل cis-regulatory من تسلسل الحمض النووي.[5][6] تستخدم MPRAs البلازميد مع cis-regulatory عناصر منظمة ليقود جينًا اصطناعيًا مثل البروتين الفلوري الأخضر. عادة ما يتم اختبار مكتبة من العناصر التنظيمية cis-regulatory باستخدام MPRAs ، يمكن أن تحتوي المكتبة من مئات إلى الآلاف من العناصر التنظيمية cis-regulatory . يتم تقييم النشاط الرقابي باستخدام نشاط مراسل المصب. يتم تقييم نشاط جميع أعضاء المكتبة بالتوازي باستخدام الرموز الشريطية لكل عنصر من عناصر رابطة cis-regulatory أحد القيود على MPRAs هو أن النشاط يفحص على البلازميد وقد لا يلتقط جميع جوانب تنظيم الجينات التي لوحظت في الجينوم.
ستار وما يليها
STARR-seq هي تقنية شبيهة ب MPRAs لفحص النشاط المحسن للفئات الجينومية المقطوعة عشوائيًا. في المنشور الأصلي، [7] وضعت فئات من جينوم ذبابة الفاكهة تم قصها بشكل عشوائي في اتجاه مجرى المفعول بالحد الأدنى. معززات المرشح بين الفئات التي تم قصها عشوائياً ستنسخ نفسها باستخدام مروج الحد الأدنى. باستخدام التسلسل كقراءة والتحكم في كميات المدخلات من كل تسلسل، يتم تقييم قوة المحسنات المفترضة بهذه الطريقة.
التشويش وما يليها
Perturb-seq الأزواج كريسبر بواسطة ضربة قاضية للجينات مع التعبير الجيني أحادي الخلية . تُستخدم النماذج الخطية لحساب تأثير ضربة قاضية لجين واحد على التعبير عن جينات متعددة.
نظام الخميرة ثنائية الهجين
يختبر فحص الخميرة الثنائية (Y2H) بروتين "الطعم" ضد العديد من البروتينات المتفاعلة المحتملة ("الفريسة") لتحديد تفاعلات البروتين_البروتين الفيزيائي. يعتمد هذا النظام على عامل النسخ، في الأصل GAL4 ، [8] كلاهما مجالان منفصلان لتنشيط النسخ والحمض النووي مطلوبان من أجل أن يتسبب البروتين في نسخ جينات المراسل . في شاشة Y2H ، يتم دمج بروتين "الطعم" في مجال الربط الخاص بـ GAL4 ، ويتم التعبير عن مكتبة من البروتينات المحتملة "التفاعلية" (المتفاعلة) في ناقل مع مجال التنشيط. في التفاعل الحي لبروتينات الطعم والفريسة في خلية الخميرة يجمع بين مجالات التنشيط والتجليد في GAL4 بشكلٍ كافٍ بحيث ينتج تعبيرًا عن جين المراسل. من الممكن أيضًا اختبار مكتبة بروتينات الطعوم بشكل منهجي ضد مكتبة من بروتينات الفرائس لتحديد جميع التفاعلات الممكنة في الخلية.
AP / MS
تنقية التقارب وطيف الكتلة (AP / MS) قادر على تحديد البروتينات التي تتفاعل مع بعضها البعض في المركبات. يُسمح لمركبات البروتينات بالتشكل حول بروتين "طعم" معين. يتم التعرف على بروتين الطعم باستخدام جسم مضاد أو علامة مؤتلفة تسمح باستخلاصه مع أي بروتينات تكونت بشكل معقد. يتم بعد ذلك هضم البروتينات إلى أجزاء قصيرة من الببتيد ويستخدم مقياس الطيف الكتلي لتحديد البروتينات بناءً على نسب الكتلة إلى الشحن لتلك الأجزاء.
المسح التحولي العميق
في المسح التحولي العميق، يتم تجميع كل تغيير محتمل للأحماض الأمينية في بروتين معين. يتم تقييم نشاط كل من هذه المتغيرات البروتينية بالتوازي باستخدام الباركود لكل متغير. بمقارنة النشاط بالبروتين من النوع البري، يتم تحديد تأثير كل طفرة. في حين أنه من الممكن اختبار كل تغيير محتمل للأحماض الأمينية بسبب التوافقيات، يصعب اختبار طفرات متزامنة أو أكثر. كما تم استخدام تجارب المسح الضوئي العميقة لاستنتاج بنية البروتين وتفاعلات بروتين البروتين.
الطفرات
يمكن التحقق من وظيفة الجينات من خلال "الجرد" المنهجي للجينات واحدًا تلو الآخر. يتم ذلك إما عن طريق الحذف أو تعطيل الوظيفة (مثل الطفرات التداخلية ) ويتم فحص الكائنات الحية الناتجة عن الأنماط الظاهرية التي توفر أدلة على وظيفة الجين المعطل.
لحمض النووي الريبي
يمكن استخدام طرق تداخل الحمض النووي الريبي (RNAi) لشل أوهدم التعبير الجيني باستخدام ~ 20 زوجًا من RNA المزدوج والذي يتم تسليمه عادةً بواسطة transfection الاصطناعية 20 ~ مير جزيئات الحمض النووي قصيرة التدخل (siRNAs) أو عن طريق الحمض النووي القصيرة المشفرة(shRNAs). يمكن استخدام شاشات RNAi ، حيث يتم اجراء فحوصات تعتمد على ثقافة الخلية أو الكائنات التجريبية (مثل C. elegans ) لتعطيل كل جين بشكل منهجي تقريبًا في جينوم أو مجموعات فرعية من الجينات (جينومات فرعية) ؛ يمكن تعيين الوظائف المحتملة للجينات المعطلة على أساس الأنماط الظاهرية المرصودة.
شاشات كريسبر
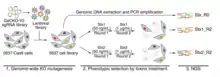
تم استخدام كريسبر-كاس 9 لحذف الجينات بطريقة متعددة في الخطوط الخلوية. تحديد كمية الحمض النووي الريبي دليل لكل الجينات قبل وبعد التجربة يمكن أن تشير نحو الجينات الأساسية. إذا قام دليل الحمض النووي الريبي (RNA) بتخريب أحد الجينات الأساسية، فسيؤدي ذلك إلى فقد تلك الخلية وبالتالي سيكون هناك استنزاف لهذا الحمض النووي الريبي المعين بعد الشاشة. في تجربة حديثة لـ CRISPR-cas9 في خطوط خلايا الثدييات، وجد أن حوالي 2000 جينة ضرورية في خطوط خلوية متعددة.[10][11] بعض هذه الجينات ضرورية في خط خلية واحد فقط. معظم الجينات هي جزء من مجمعات متعددة البروتين. يمكن استخدام هذا النهج لتحديد الفتك الاصطناعي باستخدام الخلفية الوراثية المناسبة. كريسبر و كريسبرا تتيح فقدان الوظيفة وكسب وظيفة بطريقة مماثلة. حدد كريسبري ~ 2100 جينًا أساسيًا في خط الخلايا K562.[12][13] كما تم استخدام شاشات حذف كريسبر لتحديد العناصر التنظيمية المحتملة للجينات. على سبيل المثال، تم نشر تقنية تسمى ScanDel والتي حاولت على هذه الطريقة. قام المؤلفون بحذف المناطق الموجودة خارج الجينات المسهدفة (HPRT1 المتورطة في اضطراب مندليا) في محاولة لتحديد العناصر التنظيمية لهذا الجين.[14] غاسبيريني وآخرون. لم تحدد أي عناصر تنظيمية بعيدة عن HPRT1 باستخدام هذا النهج، ولكن يمكن توسيع نطاق هذه النهج لتشمل الجينات الأخرى المسهدفة .
ملاحظات الجينوم
يمكن تحديد الجينات المفترضة عن طريق مسح الجينوم بحثًا عن المناطق التي يحتمل أن تشفر البروتينات، استنادًا إلى خصائص مثل إطارات القراءة الطويلة المفتوحة ، وتسلسلات بدء النسخ، ومواقع polyadenylation . يجب تأكيد التسلسل الذي تم تحديده على أنه جين مفترض بأدلة إضافية، مثل التشابه مع تسلسل [cDNA ] أو EST من نفس الكائن الحي، أو تشابه تسلسل البروتين المتوقع مع البروتينات المعروفة، أو الارتباط بتسلسل المروج، أو دليل على أن يتغير التسلسل المنتج عنه النمط الظاهري ويمكن ملاحظتها.
طريقة الاحجار الكريمة
طريقة الاحجار الكريمة طريقة حسابية للتنبؤ بوظيفة البروتين دي نوفو. يعتمد هذا على فرضية أن بعض البروتينات المتورطة في عملية فسيولوجية معينة قد توجد كجينين منفصلين في كائن حي وجين واحد في آخر. يتم فحص الجينوم بحثًا عن تسلسلات مستقلة في كائن ما و قراءة متاحة في كائن آخر. إذا تم دمج جينين، فمن المتوقع أن يكون لديهم وظائف بيولوجية مماثلة تجعل مثل هذا التنظيم المشترك مفيدًا.
طرق المعلوماتية الحيوية في علم الجينوم الوظيفي
نظرًا للكمية الكبيرة من البيانات التي تنتجها هذه التقنيات والرغبة في العثور على أنماط ذات معنى بيولوجيًا، تعد المعلوماتية الحيوية ضرورية لتحليل بيانات الجينوم الوظيفية. من الأمثلة على التقنيات في هذا الفصل تجميع البيانات أو تحليل المكون الرئيسي للتعلم الآلي غير الخاضع للإشراف (الكشف عن الطبقات) وكذلك الشبكات العصبية الاصطناعية أو آلات ناقلات الدعم للتعلم الآلي الخاضع للإشراف (التنبؤ الطبقي، التصنيف ). يستخدم تحليل التخصيب الوظيفي لتحديد مدى الإفراط في التعبير أو عدمه (المنظمين إيجابيين أو سلبيين في حالة شاشات RNAi) للفئات الوظيفية المتعلقة بمجموعات الخلفية. يتم توفير تحليل التخصيب الجيني القائم على تحليل الجينات بواسطة DAVID وتحليل مجموعة الجينات المخصبة (GSEA) ، [15] التحليل القائم على المسار عن طريق الإبداع [16] واستوديو المسار [17] والتحليل القائم على البروتين المعقد بواسطة كومبليت.[18]
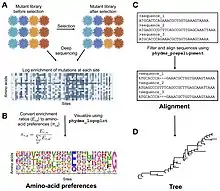
تم تطوير طرق حسابية جديدة لفهم نتائج تجربة المسح الطفري العميق. يقارن مصطلح "phydms" نتيجة تجربة المسح الطفري العميق بشجرة النشوء والتطور.[19] يسمح هذا للمستخدم بالاستنتاج إذا كانت عملية الاختيار في الطبيعة تطبق قيودًا مماثلة على البروتين كما تشير نتائج الفحص التحولي العميق. قد يسمح ذلك للمجرب بالاختيار بين ظروف تجريبية مختلفة بناءً على مدى انعكاسها على الطبيعة. كما تم استخدام المسح التحولي العميق لاستنتاج تفاعلات البروتين_البروتين.[20] استخدم المؤلفون نموذجًا ديناميكيًا حراريًا للتنبؤ بآثار الطفرات في أجزاء مختلفة من dimer. ويمكن أيضا أن تستخدم بنية طفرية عميقة لاستنتاج بنية البروتين. يمكن أن تتكون رواسب ايجابية قوية بين طفرتين في مسح طفري عميق مؤشرا على جزأين من البروتين قريبان من بعضهما البعض في مساحة ثلاثية الأبعاد. يمكن بعد ذلك استخدام هذه المعلومات لاستنتاج بنية البروتين. تم إثبات مبدأ هذا النهج من قبل مجموعتين باستخدام البروتين GB1.[21][22]
تطلبت نتائج تجارب MPRA أساليب التعلم الآلي لتفسير البيانات. تم استخدام نموذج kv-k SV-gapped لاستنتاج الكيلومترات التي يتم تخصيبها داخل متواليات cis-regulatory ذات النشاط العالي مقارنة بالتسلسلات ذات النشاط المنخفض.[23] توفر هذه النماذج قوة تنبؤية عالية. كما تم استخدام نهج التعلم العميق والنهج العشوائية للغابات لتفسير نتائج هذه التجارب عالية الأبعاد.[24] هذه النماذج في المساعدة في تطوير فهم أفضل لوظيفة الحمض النووي غير المشفرة نحو تنظيم الجينات.
تركيزمشاريع الكونسورتيوم على علم الجينوم الوظيفي
مشروع ENCODE
مشروع ENCODE (موسوعة عناصر الحمض النووي) هو تحليل متعمق للجينوم البشري الذي يهدف إلى تحديد جميع العناصر الوظيفية للحمض النووي الوراثي، في كل من المناطق المشفرة وغير المشفرة. تشمل النتائج المهمة أدلة من اغطية الجينوم على أن معظم النوكليوتيدات يتم نسخها كنصوص ترميز، أوRNAs غير مشفر، أو نصوص عشوائية، واكتشاف مواقع تنظيمية إضافية، أو مزيد من التوضيح لآليات تعديل الكروماتين.
مشروع التعبير عن الأنسجة الوراثية (GTEx)
مشروع GTEx هو مشروع وراثي بشري يهدف إلى فهم دور الاختلاف الوراثي في تشكيل التباين في النص في الأنسجة. جمع المشروع مجموعة متنوعة من عينات الأنسجة (يزيد على 50 من الأنسجة المختلفة) من أكثر من 700 متبرع بعد الوفاة. وقد أدى ذلك إلى جمع أكثر من 11000 عينة. ساعد GTEx في فهم مشاركة الأنسجة وخصوصية الأنسجة في EQTLs .[25]
مراجع
- "On the immortality of television sets: "function" in the human genome according to the evolution-free gospel of ENCODE". Genome Biology and Evolution. 5 (3): 578–90. 20 February 2013. doi:10.1093/gbe/evt028. PMID 23431001. الوسيط
|CitationClass=تم تجاهله (مساعدة) - A primer of genome science (الطبعة 3rd). Sunderland, MA: Sinauer Associates. 2004. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Bioinformatics and functional genomics (الطبعة 2nd). Hoboken, NJ: Wiley-Blackwell. 2009. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "RNA-Seq methods for transcriptome analysis". Wiley Interdisciplinary Reviews. RNA. 8 (1): e1364. January 2017. doi:10.1002/wrna.1364. PMID 27198714. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "High-throughput functional testing of ENCODE segmentation predictions". Genome Research. 24 (10): 1595–602. October 2014. doi:10.1101/gr.173518.114. PMID 25035418. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Massively parallel functional dissection of mammalian enhancers in vivo". Nature Biotechnology. 30 (3): 265–70. February 2012. doi:10.1038/nbt.2136. PMID 22371081. الوسيط
|CitationClass=تم تجاهله (مساعدة)صيانة CS1: عرض-المؤلفون (link) - "Genome-wide quantitative enhancer activity maps identified by STARR-seq". Science. 339 (6123): 1074–7. March 2013. Bibcode:2013Sci...339.1074A. doi:10.1126/science.1232542. PMID 23328393. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "A novel genetic system to detect protein-protein interactions". Nature. 340 (6230): 245–6. July 1989. Bibcode:1989Natur.340..245F. doi:10.1038/340245a0. PMID 2547163. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Genome-wide CRISPR screens for Shiga toxins and ricin reveal Golgi proteins critical for glycosylation". PLoS Biology. 16 (11). e2006951. 27 November 2018. doi:10.1371/journal.pbio.2006951. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "High-Resolution CRISPR Screens Reveal Fitness Genes and Genotype-Specific Cancer Liabilities". Cell. 163 (6): 1515–26. December 2015. doi:10.1016/j.cell.2015.11.015. PMID 26627737. الوسيط
|CitationClass=تم تجاهله (مساعدة)صيانة CS1: عرض-المؤلفون (link) - "Genome-scale CRISPR-Cas9 knockout screening in human cells". Science. 343 (6166): 84–87. January 2014. Bibcode:2014Sci...343...84S. doi:10.1126/science.1247005. PMID 24336571. الوسيط
|CitationClass=تم تجاهله (مساعدة)صيانة CS1: عرض-المؤلفون (link) - "Genome-Scale CRISPR-Mediated Control of Gene Repression and Activation". Cell. 159 (3): 647–61. October 2014. doi:10.1016/j.cell.2014.09.029. PMID 25307932. الوسيط
|CitationClass=تم تجاهله (مساعدة)صيانة CS1: عرض-المؤلفون (link) - "Compact and highly active next-generation libraries for CRISPR-mediated gene repression and activation". eLife. 5. September 2016. doi:10.7554/eLife.19760. PMID 27661255. الوسيط
|CitationClass=تم تجاهله (مساعدة)صيانة CS1: عرض-المؤلفون (link) - Gasperini, Molly; Findlay, Gregory M.; McKenna, Aaron; Milbank, Jennifer H.; Lee, Choli; Zhang, Melissa D.; Cusanovich, Darren A.; Shendure, Jay (August 2017). "CRISPR/Cas9-Mediated Scanning for Regulatory Elements Required for HPRT1 Expression via Thousands of Large, Programmed Genomic Deletions". The American Journal of Human Genetics. 101 (2): 192–205. doi:10.1016/j.ajhg.2017.06.010. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles". Proceedings of the National Academy of Sciences of the United States of America. 102 (43): 15545–50. October 2005. Bibcode:2005PNAS..10215545S. doi:10.1073/pnas.0506580102. PMID 16199517. الوسيط
|CitationClass=تم تجاهله (مساعدة)صيانة CS1: عرض-المؤلفون (link) - "Ingenuity Systems". مؤرشف من الأصل في 25 يناير 1999. اطلع عليه بتاريخ 31 ديسمبر 2007. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Ariadne Genomics: Pathway Studio". مؤرشف من الأصل في 12 فبراير 2013. اطلع عليه بتاريخ 31 ديسمبر 2007. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Protein complex-based analysis framework for high-throughput data sets". Science Signaling. 6 (264): rs5. February 2013. doi:10.1126/scisignal.2003629. PMID 23443684. مؤرشف من الأصل في 01 يونيو 2019. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "phydms: software for phylogenetic analyses informed by deep mutational scanning". PeerJ. 5: e3657. 2017. doi:10.7717/peerj.3657. PMID 28785526. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "The genetic landscape of a physical interaction". eLife. 7. April 2018. doi:10.7554/eLife.32472. PMID 29638215. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Schmiedel, Jörn M.; Lehner, Ben (17 June 2019). "Determining protein structures using deep mutagenesis". Nature Genetics. doi:10.1038/s41588-019-0431-x. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Rollins, Nathan J.; Brock, Kelly P.; Poelwijk, Frank J.; Stiffler, Michael A.; Gauthier, Nicholas P.; Sander, Chris; Marks, Debora S. (17 June 2019). "Inferring protein 3D structure from deep mutation scans". Nature Genetics. doi:10.1038/s41588-019-0432-9. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Enhanced regulatory sequence prediction using gapped k-mer features". PLoS Computational Biology. 10 (7): e1003711. July 2014. Bibcode:2014PLSCB..10E3711G. doi:10.1371/journal.pcbi.1003711. PMID 25033408. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Genome-wide prediction of cis-regulatory regions using supervised deep learning methods". BMC Bioinformatics. 19 (1): 202. May 2018. doi:10.1186/s12859-018-2187-1. PMID 29855387. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Genetic effects on gene expression across human tissues". Nature. 550 (7675): 204–213. 12 October 2017. doi:10.1038/nature24277. الوسيط
|CitationClass=تم تجاهله (مساعدة)
 صور وملفات صوتية من كومنز
صور وملفات صوتية من كومنز
 بوابة طب
بوابة طب
