ورم زوايا الفك
ورم زوايا الفك أَو مَرض الملائكية (بالإنجليزية: Cherubism) هوَ عِبارة عن مرض وراثي نِادر يُسبب بروز النصف السُفلي منَ الوجه، وَاسم المَرض مُشتق من المظهر ذو الخدين الممتلئين المشابه بِالمظهر مع الملائكة المذكورة قديماً.
| ورم زوايا الفك | |
|---|---|
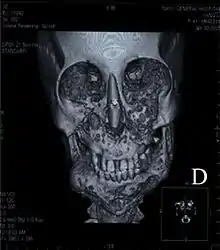 المظاهر السريرية لامرأة تبلغ 37 عاما مصابة بالمرض بدرجة خفيفة المظاهر السريرية لامرأة تبلغ 37 عاما مصابة بالمرض بدرجة خفيفة | |
| معلومات عامة | |
| الاختصاص | علم الوراثة الطبية ، وجراحة الفم والوجه والفكين ، وطب الروماتزم |
| من أنواع | مرض وراثي سائد ، وأمراض الفك ، ومتلازمة السرطان ، ومرض جلدي |
التاريخ
الملائكية تم صياغته وتوثيقه في عام 1933 بفضل الدكتور جونز في كينغستون، أونتاريو (كندا)، وقد شخّص حالة مكونة من 3 أشقاء من عائلة روسية يهودية عريقة. كان معروف عن المرض من سمات هي انتفاخ ثم الزيادة في حجم الانتفاخ ثم التراجع في هذه الآفة العظمية من الوجه[1]، ومع مرور الوقت عند وصول الأطفال عمر الخمس عشر والستة عشر والسبعة عشر التشوه الوجهي أصبح غريباً، وفي عام 1943 تم إخضاع الأطفال الثلاثة لعملية جراحية من قبل الفريق الطبي للدكتور جونز للتقليل من حجم الورم الصلب في فكوكهم، و بعد مرور أربع سنوات من الجراحة اختفى الورم و لم يظهر من جديد مرة أخرى.[2]
السمات الشكلية
المظهر العام للأشخاص المصابين بالمرض يكون بسبب فقدان عظام الفك السفلي ونتيجة لذلك يقوم الجسم بتعويض هذا النقص بنسيج ليفي، في معظم الحالات الحالة تتراجع مع نمو الطفل، ولكن في حالات نادرة الحالة تزداد سوءا وتشوه وجه المريض، والملائكية تسبب فقدان الأسنان اللبنية وعدم بزوغ الأسنان الدائمة.
إن الملائكية هي نمو شاذ نادر من العظم مورث كصفة جسمية سائدة يصيب الفكين العلوي والسفلي. تم تسجيل ما يقارب 200 حالة بالسجلات الطبية ومعظمهم من الذكور. يتم تشخيص المرض بعمر 7 سنوات ويستمر بالنمو ويمر بمرحلة البلوغ وقد يتوقف أو يزداد بالنمو مع تقدم العمر.[3]
تختلف حدة المرض من خفيف إلى حاد. والخلايا الهادمة للعظم والأخرى البانية للعظم تسهم في تحويل العظم السليم إلى نسيج ليفي وتقوم بتكوين آفات كيسية. وكما هو ملاحظ من الاسم، وجه المريض يصبح كبيرا أو غير متناسق نتيجة لتكون النسيج الليفي والتكون الشاذ للعظام، تكون العظم الإسفنجي يقود إلى الفقدان المبكر للأسنان ومشاكل في بزوغ الأسنان الدائمة، المرض يؤثر على منطقة الحجاج (منطقة في الدماغ تقع خلف العين) مسببة تغيير موقـع كرة العين نحو الأعلى وتبدو العيون مقلوبة. ويمكن أَن يكون سببه، التغيرات في الجين (SH3BP2 gene) الذي يقع في الخريطة الجينية في المكان chromosome4p16.3[4]، على الرغم من أن المرض نادر وغير مؤلم إلا أن المصاب به يعاني من اضطرابات عاطفية نتيجة التشوه.
يؤثر المرض على حركة الفكين الطبيعية والكلام. حاليا إزالة الأنسجة والعظام هي الحل العلاجي الوحيد من المرض، من المتوقع توقف نمو المرض وتراجعه ورجوع عملية البناء الطبيعي للعظام بعد مرحلة البلوغ.[5]
يحدث ورم زوايا الفك مع تطابق وراثي وعندما يُعثر على خلايا آكلة للعظم في المناطق المتأثرة في الفكين العلوي والسفلي. عدد كبير من الأكياس تتواجد مع كمية كبيرة من النسيج الليفي داخل العظم، الخيوط الليفية والأكياس تتواجد في تَرْبِيْق التابعة للنَّاتِئ الإِكْليلانيّ[6]، ذراع الفك السفلي، جسم الفك السفلي، الفك العلوي. يصاب الفك العلوي بمنطقة الحجاج العلوية وأحيانا الجزء السفلي منها. الفك العلوي وعظام الخدين تتجه للداخل وتظهر العينين كأنها محدقة للأعلى[7]، نسبة إصابة الفك العلوي أعلى في جميع الحالات مقارنة بالسفلي، في بعض الحالات يوجد أكياس تتكون في الجهة الداخلية من منطقة العين وقد يؤدي لفقدان البصر.
المسببات
المرض هو مرض وراثي سائد، والمظهر السريري يعتمد على الأليل السائد بينما الأليل الآخر الطبيعي يكون متنحي. أليل سائد واحد كافي لحدوث المرض. لأن المرض سائد المظهر السريري غير الطبيعي يتوارث إلى كل جيل على درجات مختلفة من الحدة.[5]
لذلك الآباء أو الأمهات المصابين بالمرض ينقلون النمط الظاهري لكل من الذكور والإناث. يحدث المرض نتيجة طفرة عشوائية بالجين في الأشخاص الذين لا تاريخ مرضي لهم بالعائلة. على الرغم من ذلك غير معروف لماذا يظهر المرض على الذكور أكثر من الإناث. تظهر الأعراض السريرية على المصابين بنسبة 80 إلى 100%. والأطفال يختلفون بحدة المرض بين الفكين العلوي والسفلي. يعتقد أن سبب المرض طفرة بالجين (SH3BP2). ووجد أيضا أن المرض مصاحب لاضطرابات وراثية أخرى مثل متلازمة نوونان، متلازمة رامون، ومتلازمة الصبغي س الهش.[8]
الطفرات بالجين (SH3BP2) وجدت بـ75% فقط من الحالات.[4] ونتيجة للطفرة بهذا الجين ينتج عنه فرط في إنتاج البروتين المسؤول عنه. يوجد الجين (SH3BP2) على الذراع القصيرة للكروموسوم 4 على الموقع 16.3 [4]. بروتين الجين (SH3BP2) يشارك بإرسال إشارات كيميائية لخلايا الجهاز المناعي المعروفة باسم الخلايا الأكولة الكبيرة والخلايا البائية. ما زال تأثير الطفرة الجينية بالجين (SH3BP2) تحت الدراسات، ولكن الباحثون يعتقدون أن الاضطراب في إنتاج البروتين يؤثر على الإشارات المختصة بالنسيج العظمي وبعض الخلايا المناعية، البروتينات مفرطة النشاط عادة تسبب التهابات في عظام الفك وينشط إنتاج الخلايا الهادمة للعظم، التي تقوم بتكسير النسيج العظمي أثناء عملية إعادة تشكيل العظام. الخلايا الهادمة للعظام تستشعر الزيادة في التهاب الفكين العلوي والسفلي وتتنشط أكثر لتكسير الأجزاء العظمية. فقدان العظم والالتهابات يؤدي إلى زيادة النسيج الليفي ونمو الآفات الكيسية. الإفراط في الخلايا الآكلة للعظم يساهم في تدمير العظم في الفكين العلوي والسفلي. اجتماع كلا من فقدان العظم والالتهابات عادة يؤدي إلى نمو آفات كيسية وهي من خصائص مرض الملائكية.
التشخيص
عادة التشوهات السنية تساهم في تشخيص المرض. هذه التشوهات تشمل البزوغ المبكر للأسنان اللبنية والنمو غير الطبيعي للأسنان الدائمة نتيجة تزحزحها بسبب الآفات الكيسية والآفات الاخرى. الطريقة الوحيدة لتشخيص المرض بطريقة صحيحة من خلال التحليل المتسلسل للجين (SH3BP2). وجد أن على هذا الجين طفرة مغلطة على الأكسون 9.[5] الدراسات الأولية للمريض من خلال استخدام الأشعة السينية والتصوير المقطعي المحوسب. الورم العصبي الليفي من الممكن أن يشابه الملائكية أو أن يصاحب المرض. الفحص الجيني هو الأداة التشخيصية الأخيرة.
العلاج
لأن المرض يتغير ويتطور مع مرور الوقت العلاج يجب أن يكون محددا بشكل فردي. بشكل عام الحالات المتوسطة يتم متابعتها حتى تدخل بمرحلة المرض الحادة. الحالات الحادة تتطلب تدخل جراحي لإزالة الآفات الكيسية والنمو الليفي بالفكين العلوي والسفلي. عملية نقل العظام الجراحية لعظام الوجه والفكين تكون ناجحة على بعض المرضى. الجراحة تفضل عندما يتراوح عمر المريض بين أعمار 5 و 15.[3] عند إجراء عملية على الوجه تؤخذ احتياطات بعين الاعتبار لتجنب فرع الفك السفلي الهامشي من العصب الوجهي والفرع الموجني من العصب الوجهي. الإصابة غير المقصودة للأعصاب تقلل من قوة عضلات الوجه والفك السفلي. عادة يتطلب وجود معالجة تقويمية لتجنب مشاكل الأسنان الدائمة مثل سوء الإطباق، عدم بزوغ وتغير في مكان السن الدائم [3]. المعالجة التقويمية يتم استخدامها لبزوغ الأسنان الدائمة التي تأخرت نتيجة الآفات الكيسية وغيرها في مسار البزوغ. المرضى الذين يعانون من مشاكل مثل الشفع، جحوظ العينين وفقدان البصر يلزمهم علاج لها.[3]
الوقاية
الكشف الجيني عن هذا المرض ضروري منذ أنه متصل بطفرات جينية للتقليل من حدوثه، قلة التعرف على المرض سببه اختفاء العلامات الحادة في الأبوين، الفحص قبل إنجاب الأولاد هو الوقت الملائم للتعرف على وجود مثل هذه الطفرات، الطفرة في هذا الجين تحدث بشكل عفوي ولذلك لا توجد أساليب وقاية متوفرة حاليا.
المراجع
- Jones, W. A. (1933). "Familial Multilocular Cystic Disease of the Jaws". The American Journal of Cancer. 17 (4): 946. doi:10.1158/ajc.1933.946. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Jones, W. A.; Gerrie, J.; Pritchard, J. (1950). "Cherubism--familial fibrous dysplasia of the jaws". The Journal of bone and joint surgery. British volume. 32-B (3): 334–347. PMID 14778852. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Carroll AL, Sullivan TJ (February 2001). "Orbital involvement in cherubism". Clin. Experiment. Ophthalmol. 29 (1): 38–40. doi:10.1046/j.1442-9071.2001.00363.x. PMID 11272784. مؤرشف من الأصل في 16 ديسمبر 2019. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Li CY, Yu SF (2006). "A novel mutation in the SH3BP2 gene causes cherubism: case report". BMC Med. Genet. 7: 84. doi:10.1186/1471-2350-7-84. PMC 1764878. PMID 17147794. مؤرشف من الأصل في 23 سبتمبر 2015. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Tiziani V, Reichenberger E, Buzzo CL; et al. (July 1999). "The gene for cherubism maps to chromosome 4p16". American Journal of Human Genetics. 65 (1): 158–66. doi:10.1086/302456. PMC 1378086. PMID 10364528. مؤرشف من الأصل في 16 ديسمبر 2019. الوسيط
|CitationClass=تم تجاهله (مساعدة)صيانة CS1: أسماء متعددة: قائمة المؤلفون (link) - Irfan S, Cassels-Brown A, Hayward J, Corrigan A (1997). "Orbital Cherubism". Orbit. 16: 109–112. doi:10.3109/01676839709019125. الوسيط
|CitationClass=تم تجاهله (مساعدة)صيانة CS1: أسماء متعددة: قائمة المؤلفون (link) - Emoto Y, Emoto H, Fujle W, Wakakura M (2007). "Uncorrectable Oblique Astigmatism and Impaired Binocular Vision in Case of Orbital Cherubism". Neuro-ophthalmology. 31: 191–5. doi:10.1080/01658100701648553. الوسيط
|CitationClass=تم تجاهله (مساعدة)صيانة CS1: أسماء متعددة: قائمة المؤلفون (link) - van Capelle CI, Hogeman PH, van der Sijs-Bos CJ; et al. (September 2007). "Neurofibromatosis presenting with a cherubism phenotype". Eur. J. Pediatr. 166 (9): 905–9. doi:10.1007/s00431-006-0334-6. PMID 17120035. الوسيط
|CitationClass=تم تجاهله (مساعدة)صيانة CS1: أسماء متعددة: قائمة المؤلفون (link)
 بوابة طب
بوابة طب