مرض نيمان-بيك
مرض نيمان-بيك هو مجموعة من الاضطرابات الأيضية الحادة الموروثة التي تسمح بنوع معين من الدهون بالتراكم في الخلايا. وتتراكم دهون سفينغوميالين في الليزوزومات (مرتبطة بغشاء العضيات في الخلايا). وهذه الليزوزومات تنقل بشكل طبيعي المواد عبر الخلية وخارجها. يتم التشخيص بشكل فردي ولكن قد يكون الشكل الحاد للمرض مميتا في بداية المشي اما الشكل المعتدل للمرض في بعض الحالات قد يكون طبيعياَ. يشمل هذا المرض اختلال وظيفة الايض في الشُحْمِيَّاتُ السفينغولية وهي عبارة عن دهون موجودة في أغشية الخلايا ولذلك هي تعدّ نوعا من الشُحيمات السفينغولية. في المقابل هذه الشُحيمات السفينغولية قد تم إدراجها من ضمن العائلة الكبرى لأمراض التخزين الليزوزومية.[1]
| مرض نيمان-بيك | |
|---|---|
| معلومات عامة | |
| الاختصاص | علم الغدد الصم |
| الإدارة | |
| حالات مشابهة | مرض بيك |
| التاريخ | |
| سُمي باسم | ألبرت نيمان ، ولودويغ بيك |
العلامات والأعراض
ترتبط الأعراض بالأعضاء التي يتراكم فيها السفينغوميالين. فتضخم الكبد والطحال (ضخامة الكبد والطحال) قد يسبب فقدان الشهية وانتفاخ البطن وألم. وتضخم الطحال (splenomegaly) قد يسبب أيضا بانخفاض مستويات الصفائح الدموية في الدم (الصفيحات).
ينتج عن تراكم السفينغوميالين في الجهاز العصبي المركزي (بما في المخيخ) مشية غير مستقرة (ترنح)و الإدغام في الكلام (تلعثم) وصعوبة في البلع (عسر البلع). ويسبب خلل العقد القاعدية اوضاع غير طبيعية في الأطراف والجذع والوجه (خلل التوتر). وينتج عن مرض الجزء العلوي من الجذع الدماغي ضعف في حركات العين السريعة الطوعية (الشَلَلُ فَوقَ النَووِيّ). ويتضمن امراض أكثر انتشاراَ كقشرة المخ وهياكل تحت القشرة مسببة فقدان تدريجي للقدرات الفكرية، والتي بدورها تسبب الخرف والنوبات.
والعظام يمكن ايضا ان تتأثر: وقد تشمل أعراض كتضخم تجاويف نخاع العظام ورقة العظام القشرية، أو تشوه لعظم الورك المسمى (بورك الفحجاء). ويمكن أن تحدث اضطرابات مرتبطة بالنوم مثل انقلاب النوم أي النعاس أثناء النهار واليقظة في الليل.ويمكن ايضا ان يحدث التجمد الضحكي وهو فقدان مفاجئ لقوة العضلات عند الضحك.
السبب وعلم الوراثة
حدوث الطفرات في الجين SMPD1 يسبب انواع مرض نيمان-بيك A و B, والتي تمنع الجسم من تكوين انزيم sphingomyelinase الحمضية التي تقوم بتكسير الدهون. وتسبب الطفرات في NPC1 أو NPC2 مرض نيمان-بيك نوع C (NPC) الذي يؤثر على البروتين الذي يستخدم لنقل الدهون.
ولقد تم فصل نوع D من نوع C لتحديد مجموعة المرضى الذين يعانون من اضطرابات مماثلة غير هؤلاء الذي عانوا سابقاَ من نوفا سكوتيا الشائع . ويعرف مرضى هذه المجموعة بتبادل طفرة معينة في الجين NPC1، ولذلك يستخدم NPC لكلا المجموعتين. واقترح مصطلح "نوع نيمان بيك I" و"نوع نيمان بيك II"، قبل وصف العيوب الجزيئية، للفصل بين اشكال السفينغوميالين العاليه والمنخفضه في وقت مبكر من عام 1980م.
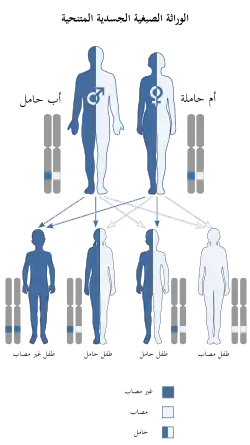
وينتقل مرض نيمان بيك بنمط وراثي متنحي والذي يعني ان كلا النسخ أو الأليلات للجين يجب ان تكون معيبة حتى تسبب المرض. وتعني"معيبة" أن هذه الجينات تغيرت بطريقة تضعف وظيفتها. في معظم الأحيان قد يحمل والدي الطفل المصابين باضطراب وراثي جسمي مقهور نسخة واحدة من جين متغير، ولكن لايتأثرون لأن النسخة الأخرى تنتج الانزيم. أما إذا كان كلا الأبوين حاملي للمرض فإن فرصة إنتاج طفل مصاب في كل حمل هي 25٪. لذلك ينصح بالاستشارة الوراثية والفحص الوراثي للعائلات التي قد تكون حاملة لمرض نيمان-بيك.
التصنيف
1. مرض نيمان بيك، المرتبط ب SMPD1 ، والذي يتضمن نوعي A و B
- مرض نيمان بيك النوع A: الطفولي الكلاسيكي.
- مرض نيمان بيك النوع B: الحشوي.
2. مرض نيمان بيك النوع C: شبه الحاد ويشمل نوعي C1 (95% من النوع C) وC2. ونوع C هو الشكل الأكثر شيوعا من المرض.[2] اما النوع C2 هو شكل نادر من هذا المرض.[3]
الإصابة
يقدر معدل الإصابة بين اليهود الأشكناز للنوع A من مرض نيمان-بيك بواحد من اصل 40,000. وتم تقدير معدل الإصابة بكلا النوعين A وB من مرض نيمان-بيك في جميع الشعوب الأخرى لتكون واحدة من اصل250,000. ويقدر معدل انتشار نوع المرض نيمان بيك C لتكون واحدة من اصل 150,000.
الفيزيولوجيا المرضية
أمراض نيمان-بيك هي مجموعة فرعية من اضطرابات تخزين الدهون والتي تسمى بالشُحيمات السفينغولية والتي تحتوي على كميات ضارة من المواد الدهنية أو الدهون، وبالتالي تتراكم في الطحال والكبد والرئتين ونخاع العظام والدماغ.
في النوع- A الطفولي الكلاسيكي البديل، تسبب الطفرة المعيبة نقص كامل في الانزيم (sphingomyelinase). والسفينغوميالين هو أحد مكونات غشاء الخلية بما في ذلك غشاء (organellar)، وبالتالي فإن نقص الإنزيم يمنع تحلل الدهون مما يؤدى إلى تراكم السفينغوميالين داخل الليزوزومات في السلائل البلعمية وحيدة الخلية. وتتوسع هذه الخلايا المتأثرة حتى يصل قطرها احياناَ إلى 90 ميكرون ثم إلى انتفاخ الليزوزومات مع السفينغوميالين والكوليسترول. وتظهرالأنسجة الضامة دهون لادن في النخاع و"منسجات البحر الأزرق" في علم الأمراض. ويتم إنشاء فجوات صغيرة عديدة من الحجم الموحد نسبيا والذي يعطي السيتوبلازم مظهر رغوي.
العلاج
لا يوجد علاج محدد لنوع A المعروف ولكن يتم التعامل مع الأعراض.
في المرضى البالغين المصابين بالنوع B، يحاول الأطباء في الحفاظ على مستويات الكوليسترول في الدم إلى مستويات طبيعية. وإذا تم استخدام الستاتين يتم مراقبة وظائف الكبد. وقد يتطلب نقل منتجات الدم في حال توسع الطحال وانخفاض مستويات الصفيحات وحدوث نوبات نزيف حادة . وإذا كان لدى المريض أعراض مرض الرئة الخلالي، فإنه قد يحتاج إلى الأكسجين.[4]
وكما تم ذكره ان محاولة زرع الأعضاء قد حققت نجاحا محدودا. وتشمل التوقعات المستقبلية استبدال للانزيم والعلاج الجيني ومحاولة زرع نخاع العظم للنوع B.
و في يناير 2009، أعلنت ACTELION انه تمت الموافقة على الدواء ميجلوستت (Zavesca) في الاتحاد الأوروبي لعلاج المظاهر العصبية التقدمية في المرضى البالغين والمرضى الأطفال مع NPC. وهذا الدواء متوفر للمرضى في الولايات المتحدة على أساس تجريبي. وفي مارس 2010، طلبت FDA معلومات اضافية قبل السريرية والسريرية التي تتعلق بـZavesca من ACTELION قبل اتخاذ قرار نهائي بشأن الموافقة على الدواء في الولايات المتحدة لNPC.
التشخيص
النوع A من مرض نيمان بيك (حوالي 85٪ من الحالات) لديه تشخيص سيء للغاية، وفي معظم الحالات يكون قاتلاَ في سن 18شهراَ. اما النوع B (في بداية الكبار) ونوع C (الطفرة التي تؤثر على جزئات مختلفة) من أمراض نيمان-بيك لديها تشخيص أفضل.
المراجع
- James, William D.; Berger, Timothy G.; et al. (2006). Andrews' Diseases of the Skin: clinical Dermatology. Saunders Elsevier. صفحة 536. ISBN 0-7216-2921-0. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Neimann-Pick Disease". Genetics Home Reference. NIH. January 2008. مؤرشف من الأصل في 24 سبتمبر 2018. اطلع عليه بتاريخ 02 أكتوبر 2012. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Ierardi-Curto, Lynne (27 December 2012). "Sphingomyelinase Deficiency Treatment & Management". Medscape Reference: Drugs, Diseases & Procedures. مؤرشف من الأصل في 22 يونيو 2018. اطلع عليه بتاريخ 22 يونيو 2013. الوسيط
|CitationClass=تم تجاهله (مساعدة);|chapter=تم تجاهله (مساعدة) - Ierardi-Curto, Lynne (27 December 2012). "Sphingomyelinase Deficiency Treatment & Management". Medscape Reference: Drugs, Diseases & Procedures. مؤرشف من الأصل في 8 أكتوبر 2017. اطلع عليه بتاريخ 22 يونيو 2013. الوسيط
|CitationClass=تم تجاهله (مساعدة);|chapter=تم تجاهله (مساعدة)
 بوابة طب
بوابة طب
