متلازمة ساثري - تشوتزين
متلازمة ساثري - تشوتزين (بالإنجليزية: Saethre–Chotzen syndrome)، كما تعرف باسم"متلازمة تسنم الرأس وارتقاق الأصابع النوع الثالث" وهو مرض خلقي نادر مرتبط بتعظم الدروز الباكر (الاطباق الغير مكتمل لبعض العظام في جمجمة الرأس). وهذا يؤثر على شكل الرأس والوجه، يؤدي إلى تكوين رأس مخروطي الشكل ووجه غير متماثل. الاشخاص المصابون بهذه المتلازمة يمتلكون جفون متدلية ويمتلكون أيضًا فراغا كبيرا بين أعينهم، وعيوبا خلقية تخلق مع الولادة في الأيدي والقدمين.[1] بالإضافة إلى هذا، الأشخاص المصابون بدرجة عالية بهذه المتلازمة يكونون مصابين بتخلف عقلي من الدرجة الخفيفة إلى المعتدلة بالإضافة إلى صعوبات التعلم، واعتمادا على شدة الإصابة، بعض الأشخاص بجاجة إلى التدخل الطبي أو الجراحي،[2] معظم المصابين يعيشون حياة طبيعية إلى حد ما.
| متلازمة ساثري - تشوتزين | |
|---|---|
| معلومات عامة | |
| الاختصاص | طب الروماتزم |
| من أنواع | تسنم الرأس وارتفاق الأصابع ، واضطراب جيني ، ومرض وراثي سائد |
| المظهر السريري | |
| الأعراض | تعظم الدروز الباكر |
الأسباب
تعظم الدروزالباكر
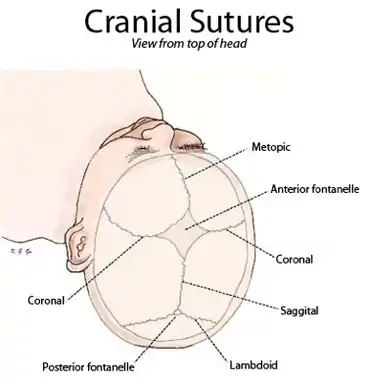
الجمجمة تتكون من ثلاثة مقاطع رئيسية تتضمن قاعدة الجمجمة(عظم القذالي)، الوجه(عظم الجبهي)، الجزء العلوي(عظام الجدارية)، جوانب الرأس (عظم الصدغي). معظم عظام الجمجمة يتم تعيينها في مكان محدد قبل الولادة. بينما، العظام الجدارية والصدغية تبقى مبتعدة عن بعضها بعض عن طريق دروز الجمجمة والتي لا تزال مفتوحة، للسماح بالتغييرات الطفيفة في شكل الرأس أثناء الولادة. الغرز الجمجمية تغلق في نهاية أول عامين بعد الولادة، بعد الانتهاء من نمو الدماغ.[1] الأشخاص الذين يعانون من هذه المتلازمة، تغلق عندهم الدروز الجمجمية (التي تفصل العظام الصدغية عن الجدارية) قبل أن يؤون الأوان " تعظم الدروز الباكر" وأحيانا يحدث هذا حتى قبل الولادة. إذا أغلقت الدروز بشكل غير متماثل أو من جانب واحد، وجهه وجبهته ستتشكل بشكل غير متساو، جنبا إلى جنب. هؤلاء المصابون يكون رأسهم مدبب الشكل لأن دماغهم نما بشكل أسرع من جماجمهم، مما أدى إلى زيادة الضغط داخل الجمجمة ويسبب بانتفاخ أعلى الرأس أو الجبهة للسماح بنمو المخ. والوجه يظهر غير متكافئ الشكل وبخاصة في مناطق العينين والخدين، ويظهر الجبين واسع وطويل القامة. بسبب الجين الغير صبغي، هنالك مساحة لملامح الوجه أن تتطور. وينتج عن ذلك أن تكون تجاويف العيون مسطحة وعظام الخدود مسطحة. وهذه التجاويف المسطحة تجعل العيون أكثر بروزا أكثر تباعدا عن بعضهما من شكلها الطبيعي. وتجاويف العيون، عظام الخد، والفك السفلي، تكون غير متطورة بسبب الوجه الأكثر تسطحا. وعلاوة على ذلك، الميل الهابط البسيط للعيون مع الأجفان المتدلية بسبب عدم التساوي العام للوجه.
علم الوراثة
 مقالة مفصلة: علم الوراثة
مقالة مفصلة: علم الوراثة

عادة يتم توريث هذا المرض باعتباره صفة سائدة جسمية. مع ذلك، في بعض الأحيان، الأطفال يحصل عندهم حذف لعدد قليل من النيوكليدات في الكروموسوم السابع على الجزء القصير أ في المنطقة 2 في المنطقة المخصصة أكثر 1, وهذا الموقع مسؤول عن الجين scs ويؤدي إلى حدوث أمراض واختلالات عصبية. وزيادة سن الوالدين قد تلعب دورا كبيرا في تطور طفرات وتشوهات جديدة.[2] كشف تحليل الارتباط وإعادة ترتيب الكروموسومات تبين أن سبب هذا المرض يكون في تشوهات في تعرجات الجين (العامل الجيني لنسح الانعوجاجات) الذي يقع على الكروموسوم السابع على الجزء القصير أ في المنطقة 2 في المنطقة المخصصة أكثر 1. جين الطي يشفر العامل الناسخ (حلزوني-عقدي-حلزوني) الذي يتحكم بتطر اللحمة المتوسطة للرأس وتكون الأنبوب الجمجمي. أكثر من 35 تشوه الانعوجاجات مختلفة يتضمن جزء ال (phlh) في البروتين بالناس المصابون بهذه المتلازمة. الطفرات تتضمن(طفرة مغلوطة، طفرة هجائية، طفرة انزياح الإطارات الحذف/الإدراج) التي إما تفصل أو تعطل جزء (phlh) في البروتين. معظم المصابين بهذه المتلازمة لديهم حذف كبير في النيوكليودات في على الكروموسوم السابع على الجزء القصير أ في المنطقة 2 في المنطقة المخصصة أكثر 1, المنطقة التي تشفر جينات النعوجاجات.[3] في البحث عن الجين المسؤول عن هذا المرض، بدأ العلماء في مركز John sHopkins Children’s Center بدراسة الجين المتخصص بالانعوجاجات و دراسة آثاره على الفئران. وهذا الجين في الفئران يؤثر على وظائف تطور عضلات وعظام الوجه والرأس واليدين والقدمين. الفئران التي كانت تفتقد نسختي هذا الجين أجهضت تلقائيا قبل الميلاد، ولديها تشوهات خطيرة في الأطراف والرأس وفشل في الأنبوب العصبي. ومع ذلك، نجت الفئران مع نسخة واحدة من هذا الجين الغير عاملة. وكشف المزيد من الفحص أن هذه الفئران لديها تشوهات طفيفة في الجمجمة والرأس والأطراف شبيهة للمصابين بمتلازمة (scs). جين الانعوجاج موجود في الفئران على الكروموسوم الثاني عشر، وفي الانسانيحتل الذراع القصير للكروموسوم الثاني عشر. مع هذه المعلومات، العلماء بدؤوا بعزل ورسم الجين المسؤول عن الانعوجاج على الذراع القصير في الكروموسوم السابع بالإنسان. وتبين لديهم أن هذا الجين في نفس الموقع يكون مفقودا بالأشخاص المصابين بهذه المتلازمة. خلال البحث في التشوهات التي قد تصيب الجين المسؤول عن الانعوجاجات، وجد أن هنالك خمسة أنواع مختلفة منها موجودة في الأشخاص المصابين بهذه المتلازمة. في حين أنه لا يوجد أي نوع من هذه التشوهات في الأشخاص السليمين، هذا يقدم دليل دليل قوي أن هذا الجين هو المسؤول عن الإصابة بمرض (scs1). الباحثون أيضا قاموا بدراسة هذا الجين في ذبابة الفاكهة من أجل تحديد وظيفتها. اكتشفوا أن وجود جزيئين من بروتينات الانعوجاج متصلين ببعضهم، ووظيفة جين النعوجاج كعامل نسخ DNA, هذا يعني أنه يرتبط بموقع معين على جريء DNA حتى يسيطر على الجينات التي هي قيد التشغيل أو مفعلة. الغالبية العظمى من الطفرات في هذه الجينات تتداخل مع كيفية تداخل البروتين مع جزيء ال DNA ومنع تفعيل الجينات الأخرى التي عادة ما يتم تفعيلها أثناء التطور الجيني.[1]
علامات وأعراض
الأشخاص المصابون بهذه المتلازمة يتأثرون بشكل مختلف. حتى ضمن العائلة الواحدة، ويمتلك الأفراد المتضررين ملامح وخصائص مختلفة. غالبية الأشخاص الذين يعانون SCS يتأثرون بشكل معتدل، مع ملامح الوجه غير المتكافئة، ووجه مسطح عظام الخد، والفك السفلي. بالإضافة إلى تشوهات جسدية، والأشخاص المصابون يعانون أيضا من تأخيرات في النمو، مما يؤدي إلى قصر القامة نسبيا. على الرغم من أن معظم الأفراد مع SCS هم من أصحاب الذكاء العادي، قد يكون بعض الأفراد لديهم درجات خفيفة إلى معتدلة من التخلف العقلي. أشد حالات هذه المتلازمة، ترافقها تشوهات الوجه العالية الخطورة، تحدث عندما تغلق الغرز الجمجمة متعددة قبل الأوان.[1]
- علامات الجمجمة

- عيوب في اليدين والقدمين
- عيوب العين

- عيون موضوعة بشكل غير متساو مما يؤدي إلى تداخلها (الحول) أوابتعادها عن بعض (فرط)[5]
- مشاكل في الرؤية بسبب التشوهات في خلقية الوجه، والذي يسبب الاضطرابات في توزيع عضلات العين، مما أدى إلى الحول (عبرت عيون)[2]
- تضيق في قنوات المسؤولة عن تسييل الدموع (تضيق القناة المسيل للدموع)[2]
- الجفون المتدلية (إطراق)[2]
- هبوط في شقوق الجفون المتدلية (الفصل بين الجفون العليا والسفلى)[2]
- قصر النظر (قصر النظر)[4]
- تتطعجات في طبقات الجلد الموجودة في منطقة الجفن مما يؤدي إلى تغطية الزاية الداخلية للعين[3]
- تضيق الأجفان (إطراق الثنائي مع انخفاض حجم الجفن)[3]
- ضمور العصب البصري[3]
- أخطاء حرارية[3]
- عيوب في الأنف والأذن والحنجرة
- أذنين صغيرتين ومستواهما منخفض ومن الممكن أن يحصل لهما استدارة للخلف[4]
- انعوجاج في الأنف (قمة الانف تكون منحرفة للأسفل ) انحراف الحاجز الأنفي[1]
- سوء الإطباق في الأسنان و هذه الظاهرة ترتبط مع تشوهات في الأسنان تتضمن نقص تصبغ مينا (المينا رقيقة بسبب عدم اكتمال تشكيلها )، زيادة عدد الاسنان عن العدد الطبيعي (الأسنان إضافية)، (والأسنان على شكل غير طبيعي صغيرة)[3]
- الحنك المشقوق مرتفع المدى [3]
- عيوب أقل شيوعا :
التشخيص
تشخيص ما قبل الولادة
التشخيص قبل الولادة لهذه المتلازمة تكون في غاية الخطورة خلال فترة الحمل، ولكن من النادر جدا تأدية هذا النوع من التشخيص. وعلاوة على ذلك، فهذه الطريقة الوحيدة للتشخيص إذا الطفرة المسببة للمرض تم تحديدها. هناك عدد من التقنيات المختلفة التي اختبارات ما قبل الولادة يمكن القيام بها. عادة ما يتم إجراء اختبارات ما قبل الولادة في الاسابيع (15-18) من الحمل، وذلك باستخدام بزل السلى لاستخلاص الحمض النووي DNA من خلايا الجنين. ويمكن أيضًا إجراء هذه الاختبار خلال الاسابيع (10-12) من الحمل باستخدام عينات من الزغابات المشيمائية (CVS) لاستخراج الحمض النووي من الجنين.[6] في الآونة الأخيرة، كان هناك اهتمام متزايد في استخدام معدات الموجات فوق الصوتية من أجل الكشف عن تشوهات الجمجمة الجنين الناتجة عن الاندماج الغير ناضج لعظام الجمجمة .[4] التشخيص السريري ويعتمد التشخيص الشامل لهذه المتلازمة في المقام الأول على النتائج السريرية، والملاحظات بناء على فحص dysmorphology (تقييم العيوب الهيكلية) والتقييم الشعاعي (الأشعة السينية، بمسح الرنين المغناطيسي، ct ) ويستند التشخيص السريري عموما على وجود الخصائص التالية:[6]
- تعظم الدروز الباكر
- النتيجة الأكثر شيوعا للالتحام المبكر لعظام الجمجمة،[6] وعادة ما **يصاحب تعظم الدروز الباكر شكل غريب للجمجمة (على سبيل المثال، قصر الرأس [قصير وعريض ] وتسنم الرأس [مخروطي الشكل ]).[6]
- عند تحديد ما إذا كان الفرد مصاب بهذه المتلازمة، يقوم الطبيب بفحص جمجمة المريض وشكلها وسوف تكون كافية على معرفة ما إذا كان قد حدث التحام سابق لأوانه استنادا إلى شكل الجمجمة.[6]
- خط بداية الشعر يكون منخفض، وعدم تناسق الوجه، تدلى الجفون، والحول (كسل في العيون)[6]
- آذان صغيرة مع انتفاخ واضح لصيوان الأذن[6]
- تشوهات في الأطراف بما في ذلك الأورام (إبهام القدم )، قصر الاطراف، وأصابع وتراء جزئيا، يكون الإبهام الكبير للقدم على شكل الكردوس الثلاثي (اصبع القدم الكبير)، وتكرار السلامية (احدى عظام في اصبع القدم ) البعيدة في ابهام القدم[6]
التشخيص الجزيئي والوراثي
التشخيص السريري للSCS يمكن التحقق منه عن طريق اختبار الجينات TWIST1 (الجينات فقط التي تسبب الطفرات التي تتسبب SCS) للطفرات باستخدام تحليل الحمض النووي، مثل تحليل التسلسل، حذف / تحليل التضاعف، وعلم الوراثة الخلوية / تحليل FISH. تحليل تسلسل اكسون 1 (منطقة الترميزTWIST1 ) يوفر وسيلة جيدة للكشف عن وتيرة الطفرات في الجين TWIST1. وتشمل هذه الطفرات missense، nonsense، splice site، والحذف داخل الجين / الإدراج. ويحدد تحليل حذف / تضاعف الطفرات في الجين TWIST1 التي لم يتم الكشف بسهولة عن طريق تحليل التسلسل. وتشمل الطرق الشائعة PCR، MLPA، وميكروأري الكروموسومات (CMA). التحليل الوراثي الخلوي / FISH يربط علامات الحمض النووي الملونة للكروموسوم المتمسخ ومن ثم يتم فحصها تحت إضاءة الفلورسنت، والذي يكشف عن الطفرات التي تسببها نقل المواقع أو العكس في 7p21. في بعض الأحيان، والأفراد مع SCS لديهم كروموسوم النقل، الانعكاس، أو حلقة كروموسوم 7 متضمنا 7p21 مما أدى إلى نتائج شاذة، مثل زيادة تأخر في النمو.[6] الأفراد مع SCS، عادة ما يكون لديهم عمل الدماغ طبيعي، ونادرا ما يعانون من مشاكل عقلية. لهذا السبب، إذا كان الفرد لديه SCS وتخلف عقلي معا، ينبغي أن يخضع الجين TWIST1 على فحص أكثر دقة لأن هذه ليست سمة طبيعية لل SCS . [1] الاختبار الوراثي الخلوي والاختبار الجيني المباشر يمكن أن تستخدم أيضا لدراسة عيوب الجينات / الكروموسومات. الاختبار الوراثي الخلوي هو دراسة للكروموسومات للكشف عن الزيادة أو النقص من الكروموسومات أو قطاعات الكروموسوم باستخدام فلوري التهجين في الموقع (FISH) و / أو التهجين الجينومي المقارن . اختبار الجينات المباشر يستخدم الدم، والشعر، والجلد، السائل الذي يحيط بالجنين، أو الأنسجة الأخرى من أجل إيجاد الاضطرابات الوراثية. اختبار الجينات المباشر يحدد ما إذا كان الفرد لديه SCS عن طريق اختبار الدم للطفرات في الجينTWIST [6] التشخيص التفريقي: (من الحالات الأكثر شيوعا بالخطا) حتى وقت قريب، كثيرا ما اختلف الخبراء حول ما إذا كان المريض عنده SCS، متلازمة كروزون، تعظم الدروز الباكر المعزول، أو بعض الأمراض الأخرى لأن أعراض ترتبط بشكل وثيق، لم يكن لديهم أي وسيلة للتمييز بين كل منها. ومع ذلك، لدينا الآن اختبار الجينات المباشر، والذي يسمح لتشخيص أكثر دقة لأنه يسمح لها أن تكون متمايزة عن بعضها البعض على أساس الجين المتحور في كل حالة.[1] وفيما يلي قائمة من الشروط الخلط عادة / تشخص خطأ لSCS، بعض من أعراضها، والذي تحور الجين كل تتضمن ما يلي:
| المرض\الحالة | الأعراض | الجين المتحور |
|---|---|---|
| SCS | عيون متباعدة على نطاق واسع، ضبط شعري منخفض، عيون منخقضة، بين الأصابع حزام، وآذان مشوهة، عيون عابرة، و هبوط مائل لشقوق الجفن | TWIST1 |
| Robinow-Sorauf Syndrome | عيون متباعدة، الحاجزمنحرق، الجمجمة الخلفية مسطحة، وآذان مشوهة، عيون عابرة، جاحظ الفك، والازدواجية في السلامية البعيدة | TWIST |
| Muenke Syndrome | عيون متباعدة على نطاق واسع، الراس متضخم، فقدان السمع، والخدين مسطحة، وآذان منخفضة | FGFR3 |
| Crouzon Syndrome | عيون متباعدة على نطاق واسع، حد الرأس قصير، فقدان السمع، والعيون منتفخة، الأنف على شكل منقاروالأذنين منخفضة، الحول، جاحظ الذقن، والعضد وعظم الفخذ قصير | FGFR2 & FGFR3 |
| Pfeiffer Syndrome | عيون متباعدة على نطاق واسع، الفك غير متطور بالكامل، الأنف على شكل أنف، وفقدان السمع، والعيون المنتفخة | FGFR1 & FGFR2 |
| Apert Syndrome | عيون متباعدة على نطاق واسع، جبهته بارزة، الجمجمة الخلفية مسطحة، والعيون منتفخة، الأذنين منخفضة والوجه مسطح أو مقعر، الإبهام قصير، وأصابع مكففة | FGFR2 |
| Isolated Unilateral Coronal Synostosis | التشوه الوحيد هو اندماج سابق لأوانه من الغرز. إذا تركت دون علاج، يمكن أن يؤدي إلى عدم تناسق في SCS الوجه | FGFR (any) |
| Baller-Gerold Syndrome (BGS) | حد الرأس قصير والعينين منتفخة، الجبين مسطح، تبكل، شعاعي التشوه مع تخفيض عدد الأرقام، الإبهام متخلف أو مفقود والكعبرة، وتأخر النمو | RECQL4 |
الإجراءات والعلاج
التشوهات الجسدية الناجمة عن SCS خفيفة عادة ولا تتطلب سوى إجراء عمليات جراحية بسيطة أو لا أي إجراء على الإطلاق. واحدة من الأعراض الشائعة لSCS هو تطوير القصير (قصر الأصابع)، وأصابع اليدين والقدمين واسعة الغشاء (ارتفاق الأصابع). هذه الخصائص لا تسبب أي مشاكل لوظيفة اليدين أو القدمين، وبالتالي، لا يلزم إجراء طبي لعلاج هذه التشوهات، ما لم يطلب المريض ذلك. ربط الأصابع قد يؤثر على قاعدة الأصابع، مما أدى إلى تأخرنمو في اليد خلال مرحلة الطفولة، ولكن هذا لا يسبب إعاقات وظيفية. في بعض الأحيان، الأفراد مع SCS يطورون أصابع واسعة لأن العظام في نهايات أصابع القدم تكرر نفسها. ويرى هذا خصوصا في إصبع القدم الكبير، ولكن لا يتطلب أي تدخل جراحي لأنه لا يؤثر سلبا على وظيفة القدم الكلية. الأفراد مع هذه التشوهات يمكنهم المشي بشكل طبيعي وارتداء الأحذية العادية.[8]
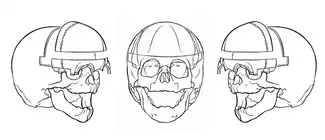
في الحالات الأكثر شدة، يطلب عمليات جراحية متكررة ومراقبة سريرية أثناء التطور. وينبغي أن يكون الطفل المولود مع التحام العظام الاكليلية من جانب واحد غير متكافئ أن يخضع لرأب القحف داخل السنة الأولى من الحياة للحيلولة دون زيادة الضغط داخل الجمجمة، ومنع عدم تناسق الوجه التدريجي. رأب القحف هو إجراء جراحي لتصحيح اندماج عظام الجمجمة المبكر. تعمل العملية الجراحية على إعادة بناء العظام والدروز من أجل تعزيز النمو الطبيعي.[3] رأب القحف هو ضروري من أجل مواصلة النمو ومهم لسببين رئيسيين. أولا وقبل كل شيء، يجب على الجمجمة أن تكون قادرة على استيعاب تنامي الدماغ بعد الولادة ، التي لا يمكن لأن الجمجمة لا تنمو بسرعة نموالمخ طالما لا تزال الغرز تندمج. وهذا يؤدي إلى زيادة في الضغط المحيط بالدماغ ويمنع المخ من النمو، مما تسبب للشخص لتجربة مشاكل كبيرة، وإذا تركت دون علاج يمكن أن يؤدي في نهاية المطاف إلى الموت. ثانيا، قد تكون هناك حاجة لرأب القحف لأغراض المظهر.[6] وهذا هو الحال في الأفراد مع التحام العظام الإكليلية من جانب واحد غير المتكافئة، الأمر الذي يتطلب جراحة ترميمية للوجه والجمجمة خاصة. إذا لم يتم تنفيذ رأب القحف، وخاصة في الأشخاص الذين يعانون التحام العظام الإكليلية من جانب واحد، عدم تناسق الوجه سيزداد سوءا مع مرور الوقت، وهذا هو السبب لأن رأب القحف ينبغي أن يطبق في أقرب وقت ممكن.[8]
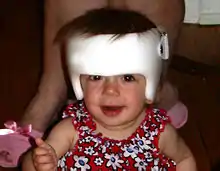
قد تكون هناك حاجة أيضا لعملية جراحية في الأشخاص الذين يعانون مشاكل في الرؤية. تنشأ مشاكل في الرؤية عادة بسبب عدم وجود مساحة في مدار العين والجمجمة بسبب بنية العظام غير الطبيعية في الوجه. تقليل المساحة قد يؤدي أيضا إلى قنوات دمعية غير طبيعية أو مفقودة وتلف الأعصاب. وعادة ما يتطلب جراحة من أجل زيادة مساحة الجمجمة، تصحيح قنوات الدمع / أو تصحيح انخفاض الجفون للحيلولة دون الحول (العين الكسولة).[1] قد تكون هناك حاجة أيضا لعملية جراحية Midfacial خلال مرحلة الطفولة المبكرة لتصحيح مشاكل في الجهاز التنفسي، سوء الإطباق في الأسنان، وصعوبات في البلع. يتم تصحيح الحنك أيضا مع الجراحة، ويمكن أن تتضمن استخدام أنابيب فغر الطبلة. إذا لزم الأمر، سوف يتلقى الفرد العلاج التقويمي و / أو المعالجة التقويمية بعد تطوير الوجه الكامل.[1] في حين ان فقدان السمع كثيرا ما يتم ربطه بال SCS، فمن المستحسن أن فحص السمع يستمر طوال مرحلة الطفولة.[3] بعد جراحة في الجمجمة، قد تكون هناك حاجة للطفل على ارتداء خوذة أو أي شكل آخر من أشكال الحماية رأسه حتى عظام الجمجمة ترجع إلى مكانها. هذا يستغرق عادة حوالي ثلاثة أشهر ويعتمد على عمر الطفل وشدة الحالة. بعد العلاج، الأفراد مع SCS يبدون و يتصرفون بشكل طبيعي تماما، لذلك لا يمكن لأحد حتى يكون قادرا على أن أقول أن لديهم SCS.[9]
الانتشار
SCS هو متلازمة تعظم الدروز الباكر الأكثر شيوعا ويصيب 1 من كل 25000 إلى 50000 الأفراد.[8] ويحدث في جميع المجموعات العرقية والإثنية، ويصيب الذكور والإناث على حد سواء.[1] إذا كان يحمل أحد الوالدين على نسخة من تحور الجين SCS هناك فرصة 50% أن أطفالهم سيحملون طفرة جينية أيضًا، وفي هذه الحالة، فإن الطفل قد تظهر أو لا تظهر عليه علامات SCS. وهناك أيضا فرصة 50٪ أن أطفالهم لديهم نسختان من الجين العامل، وسيكون لذلك، ليس لديهم SCS. إذا كان كلا الوالدين يحمل نسخة واحدة من تحور الجين SCS، هناك فرصة 25٪ أن أطفالهم لديهم نسختين تحور الجين (حتى الطفل سيتطور SCS شديد)، فرصة 25٪ أن أطفالهم لديهم نسختين عاديتين لل الجينات (ما من شأنه أن تكون طبيعية تماما)، وفرصة 50٪ طفلهما سيحمل أحد الجينات نسخة طفرة و1 نسخة عادية (بحيث يمكن أن يظهر طفل أو قد لا يظهر ال SCS . [1] وفي حالات نادرة، اثنين من الآباء الطبيعيين يمكن أن يكون له طفل مع SCS بسبب طفرة دي نوفو. السبب الدقيق لطفرة الدي نوفو غير معروف، ولكن لا يبدو أن له صلة إلى أي شيء الوالدين فعلا أو لم يفعلا أثناء الحمل.[10] SCS بسبب طفرة دي نوفو من النادر جدا في نسبة الحالات السابقة غير معروف.[6]
التاريخ
في عام 1931، هاكون سايثر، وهو طبيب نفسي نرويجي، وصف خصائص مماثلة بين الأم وابنتيها. كان لديهم ميزات طويلة ومتفاوتة في الوجه، وخطوط الشعر منخفضة، أصابع قصيرة، وربط بين الإصبع الثاني والثالث وبين الثانية والثالثة، والرابعة من أصابع القدم. وبعد مرور عام في عام 1932، F. Chotzen، وهو طبيب نفسي ألماني، وصف أب وابنيه لديهما خصائص مشابهة جدا للأم وبناتها، فضلا عن وجود فقدان السمع، قصر القامة، والتخلف العقلي البسيط. وبالتالي، تم اشتقاق اسم متلازمة Saethre-Chotzen من اثنين من العلماء، الذين وصفا الحالة بشكل منفصل دون أي معرفة سابقة ببعضهما.[1]
المراجع
- Blanchford, Stacey L (2002). The Gale Encyclopedia of Genetic Disorders. Michigan: Gale Group. صفحات 1019–1021. ISBN 9780787656140. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Allanson, Judith, Cassidy, Suzanne (2010). Management of Genetic Syndromes. New Jersey: John Wiley & Sons, Inc. صفحات 230–235. ISBN 9780470191415. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Clauser L, Galie M. "Saethre-Chotzen Syndrome" (PDF). Orphanet. مؤرشف من الأصل (PDF) في 26 سبتمبر 2018. اطلع عليه بتاريخ 28 أكتوبر 2012. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Wynbrandt, James (2008). Genetic Disorders and Birth Defects. New York: Facts on File, Inc. صفحات 340. ISBN 9780816063963. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Saethre-Chotzen Syndrome". International Craniofacial Institute. مؤرشف من الأصل في 09 يونيو 2015. اطلع عليه بتاريخ 28 أكتوبر 2012. الوسيط
|CitationClass=تم تجاهله (مساعدة); تحقق من التاريخ في:|تاريخ أرشيف=(مساعدة) - Gallagher E, Ratisoontorn C, Cunningham M. "Saethre Chotzen Syndrome". NCBI. مؤرشف من الأصل في 03 فبراير 2016. اطلع عليه بتاريخ 28 أكتوبر 2012. الوسيط
|CitationClass=تم تجاهله (مساعدة); تحقق من التاريخ في:|تاريخ أرشيف=(مساعدة) - "Saethre-Chotzen Syndrome". Children's Hospital and Medical Center. مؤرشف من الأصل في 07 سبتمبر 2015. اطلع عليه بتاريخ 25 أكتوبر 2012. الوسيط
|CitationClass=تم تجاهله (مساعدة); تحقق من التاريخ في:|تاريخ أرشيف=(مساعدة) - "Saethre-Chotzen Syndrome". Boston Children's Hospital. مؤرشف من الأصل في 16 سبتمبر 2013. اطلع عليه بتاريخ 28 نوفمبر 2012. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Surgical Options for Craniosynostosis". Johns Hopkins Medicine. مؤرشف من الأصل في 07 ديسمبر 2013. اطلع عليه بتاريخ 28 نوفمبر 2012. الوسيط
|CitationClass=تم تجاهله (مساعدة); تحقق من التاريخ في:|تاريخ أرشيف=(مساعدة) - "Saethre-Chotzen Syndrome". Seattle Children's Hospital. مؤرشف من الأصل في 23 يونيو 2016. اطلع عليه بتاريخ 28 نوفمبر 2012. الوسيط
|CitationClass=تم تجاهله (مساعدة)
 بوابة طب
بوابة طب