متلازمة زيلويغر
متلازمة زيلويغر (أو المتلازمة المخية الكبدية الكلوية) هو اضطراب خلقي نادر يتميز بنقص في أو غياب كامل للبيروكسيسومات الفعالة في خلايا المريض.[1] يعد أحد الاضطرابات من عائلة اضطرابات طيف زلويغر، والتي تعد أمراض مسببة لحثل المادة البيضاء في الدماغ. سميت هذه المتلازمة نسبة إلى هانز زلويغر (Hans Zellweger)، برفيسور في طب الأطفال وعلم الجينات في جامعة ايوا (University of Iowa)، والذي قام بالبحث في هذا المرض.[2][3]
| Zellweger syndrome | |
|---|---|
| معلومات عامة | |
| الاختصاص | علم الوراثة الطبية |
البيروكسيسومات
البيروكسيسومات هي عضيات مغلفة بأغشية، متواجدة في جميع الخلايا حقيقية النوى. تنتج البيروكسيسومات عن طريق الانقسام أو تصنع في الشبكة الإندوبلازمية. تحتوي البيروكسيسومات على 70 نوع مختلف من الإنزيمات اللازمة للقيام بعمليات الأيض الطبيعية الخاصة بالدهون، وأيضا القيام بالعمليات الحيوية الضرورية للمحافظة على صحة الإنسان ونموه الطبيعي.[4]
أعراض المرض وأثره على أجهزة الجسم وإمكانية التحكم بالأعراض
متلازمة زيلويغر هي واحدة من ثلاثة اضطرابات في الجسيمات التأكسدية[5]، الأول هو طيف زيلويغر للاضطراب الحيوي الجسيمي التأكسدي (PBD-ZSD)، الثاني هو اضطراب في حثل المادة البيضاء الكظري للمواليد الجدد (NALD)، والثالث داء ريفسام الخاص بالرضع[6][7] (IRD)، وعلى الرغم من كون الاضطرابات الثلاثة تشترك في كونها تتشابه في الخصائص الجزيئية البيولوجية، إلا أن متلازمة زيلويغر هي أشدها وأكثرها حدة من بين الثلاثة.[8]
تسبب متلازمة زلويغر عدة تشوهات ظاهرية نتيجة لأثر المرض على العظام ونمو المريض. تتضمن هذه التشوهات أذنين غير متماثلتين، تشوه في شحمة الأذنين، زيادة في طيات جلد الرقبة، تجعد في جفون العينين، انحراف العينين إلى أعلى، وجه مسطح المظهر، وعلو الجبهة، وغيرها من التشوهات في منطقة الرأس والوجه للمريض.[9]
متلازمة زيلويغر لها علاقة مع اختلال في السيالات العصبية، وتحديد المواقع العصبية، ونمو المخ[5]، وقد تظهر انخفاضا في ميالين الجهاز العصبي المركزي وتحديدا المخ والمسؤولة عن المحافظة على الوظائف الطبيعية لدى الجهاز المركزي العصبي. المصابون بالمتلازمة قد يظهرون انحلال متقدم للأعصاب المسؤولة عن الحس في الدماغ والذي بدوره يؤدي إلى فقدان مستمر للسمع والرؤية.[5]
تؤثر متلازمة زلويغر على وظائف عدة أجهزة أخرى في الجسم:-
البصر
فقدان النظر شائع بين مرضى متلازمة زلويغر الناجم عن ضمور شبكية العين وتشوهات العصب البصري، لذلك مراجعات بصرية دورية مطلوبة للمرضى. على الرغم من أن إعتام عدسة العين (الماء الأبيض) نادر الحدوث، ولكن في حال حدوثه، فإن علاجه في عمر مبكر قد يحافظ على الرؤية، مع العلم أن هذا قد يتطور إلى عدم فعالية الشبكية. يمكن استخدام النظارات الطبية عند الحاجة إليها لإصلاح الانعكاسات الخاطئة للأشعة المرئية. في الأطفال المصابين بمتلازمة زلويغر، لا يوجد داعي لأكثر من تخطيط كهربائي للعين لتقييم حالة العين، حيث أنه تم إثبات عدم قدرة هذا الفحص على توقع حالة البصر وتقدم الحالة. التصوير المقطعي للتماس البصري قد أثبت فعاليته في تقييم حالة الشبكية للأطفال المصابين بالمرض عند الأطفال القادرين على الاستجابة لأوامر الطبيب خلال النظر إلى مصدر ضوء.[4]
السمع
يتفاوت الكثير من مرضى متلازمة زلويغر بأثر المرض على حاسة السمع، لذلك يجب تقييم قدرة السمع لدى الأطفال المصابين بالمرض سنويا، واستخدام الالات التي تساعد على السمع عند الأطفال الذين وجد لديهم ضعف بالسمع. في حالة ضعف حاد في السمع وعدم قدرة الالات المساعدة على تحسين السمع لدي الطفل المصاب، يتم زرع جهاز كوتشلير لتحسين حاسة السمع لديهم.[4]
العظم والأسنان
قد يعاني الأطفال المصابون بمتلازمة زلويغر بشكل حاد بخلل التنسج الغضروفي المنقط (chondrodysplasia puntata)، حيث يتسبب هذا المرض بتنقير نهايات العظام الطويلة وتغيرات في الهيكل العظمي. قلة كثافة العظام التي تسوء مع الزمن تكون جزء من متلازمة زلويغر معتدل أو ضعيف الشدة، وقد حصلت الكسور المرضية عند بعض المرضى بدون دلائل على حوادث أو إصابات. عند الأطفال بعمر عام أو أكثر الذين لا يملكون القدرة على تحمل الأوزان أو لديهم ماض بكسور في العظام فيجب الأخذ بالاعتبار تقييم حالة العظام في الجسم، وذلك يشمل أشعة x ذات الطاقة المضاعفة وفحص فيتامين د. يفضل فحص أسنان المرضى كل ستة شهور حيث أن المرضى يعانون من تشوهات في مينا الأسنان الدائمة، وعليه يجب أخذ العناية الضرورية للأسنان.[4]
الوظائف العصبية
لقد تم اكتشاف حالات من الصرع عند معظم الأطفال الرضع الذين يعانون من المرض بحالة حادة، بالإضافة إلى 23٪ من الأطفال الذين يعانون من المرض الأقل حدة أوالمتوسط. بإمكان فحص الرسم الكهربائي للدماغ تحديد استمرارية ومدة نوبات الصرع لدى المرضى، ويجب القيام به في الحالات المتوقع فيها تغير النوبات. من الأدوية المستخدمة للتحكم بنوبات الصرع لدى المرضى الفينوباربيتال وكلونازيبام وتوبيرامات.[4]
قد يتطور لدى المرضى مرض حثل المادة البيضاء والذي قد يكون صامت أو متقدم. لذلك ينصح بعمل (MRI) للدماغ مصحوبة بالدراسات الضرورية الأخرى إذا توقعت ذلك عيادة المريض. إن تغير المادة البيضاء لدى المريض بإمكانه تبديل الحالة السلوكية للمريض وقدرته على التحكم بحركات الجسم الارادية.[4]
ينصح بتقييم قدرات المرضى الحركية والكلامية في سن صغير وتقديم التدخلات اللازمة باكرا.[4]
الكبد
لدعم وظائف الكبد، ينصح بإعطاء فيتامين ك كمدعم بجرعة 2.5 – 5 ملغم يوميا. متلازمة زلويغر تؤثر أيضا على عمليات الأيض للأحماض المرارية، لذا يمكن تحسين وظائف الكبد عن طريق علاج الأحماض المرارية الأساسي (حمض الكوليك وحمض الكينودياوكسيكوليك)، الذي يقلل من تراكم طلائع الأحماض المرارية، كDHCA وTHCA. في الوقت الحالي، الدراسات المتوفرة التي تقيم فعالية علاج الأحماض المرارية في متلازمة زلويغر وأمراض البيروكسيسومات المشابهة له محدودة وتعطي نتائج مختلفة اعتمادا على حدة المرض. يجب أيضا فحص ومتابعة عوامل التخثر ووظائف الكبد التركيبية المختلفة. خلل الكبد الوظيفي قد يؤدي إلى دوالي تستجيب للعلاج المناسب لها.[4]
الغدة الكظرية
كما في اضطرابات البيروكسيسومات الأخرى، فإن قصور الغدة الكظرية الأساسي قد يحدث في متلازمة زلويغر. ينصح بأن يتم إجراء فحوصات سنوية، أو فحوصات أكثر تكرارا، للغدة الكظرية وهرموناتها (الهرمون الموجه لقشر الكظر ACTH وفحص الكورتيزول الصباحي) لمرضى زلويغر الذين تخطوا سن العام. في حالة وجود قصور الغدة الكظرية، يتم بدء علاج الاستبدال الكظري بالجرعات الاعتيادية. يجب أيضا تحضير جرعات أخرى مناسبة أكثر قوة في حالة مرض أو حمى حادة مفاجئة، أو في حالة اضطر المريض لإجراء عملية جراحية.[4]
الكلى
إن الأطفال أكبر من سن 4 إلى 6 سنين المصابون بمتلازمة زلويغر يجب مراقبتهم من مرض تكون البلورات في البول (hyperoxaluria) الذي قد يؤدي إلى تكون حصوات وفشل كلوي. بالإمكان مراقبة ذلك عن طريق فحص حمض الاوكزاليت وفحص الكيرياتين في البول. بالإمكان استخدام جهاز الأشعة فوق الصوتية للفحص في حال وجود حصوات.[4]
السبب
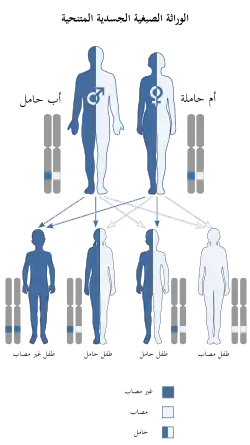
متلازمة زلويغر هي اضطراب ناتج عن طفرة متوارثة كصفة متنحية، في جينات البيروكسينات، وهي بروتينات ضرورية للتجميع الطبيعي للبيروكسيسومات. في العادة تكون هذه الطفرة لدى المرضى في جينات PEX1، PEX2، PEX3، PEX5، PEX6، PEX10، PEX12، PEX13، PEX14، PEX16، PEX19، وPEX26.[10] في أغلب الحالات، تقوم الطفرات لدى المرضى بتعطيل أو تقليل فعالية كلتا النسختين من جينات PEX من جهة الأب ومن جهة الأم بصورة كبيرة.
كنتيجة لتعطيل فعالية البيروكسيسومات، تتراكم الأحماض الأمينية الطويلة جدا والأحماض الأمينية متفرعة السلاسل في أنسجة وخلايا الشخص المصاب، حيث يتم في الوضع الطبيعي تحليل هذه الأحماض الأمينية في البيروكسيسومات. يؤدي تراكم هذه الليبيدات إلى تعطيل الوظائف الطبيعية لعدة أجهزة في الجسم. بالإضافة إلى ذلك، قد يظهر المصابون بهذا المرض مستويات منخفضة من البلازمولوجينات، وهي ليبيدات مفسفرة تحتوي على مجموعة إيثر ضرورية بالأخص في وظائف الدماغ والرئتين.
التشخيص
بالإضافة إلى الفحوصات الجينية التي تتعلق بجينات PEX [11][12]، فهناك فحوصات بالكيمياء الحيوية أثبتت فعاليتها بتشخيص متلازمة زلويغر وأمراض اخرى تتعلق بالبيروكسيسومات. عادة فان المصابين بمتلازمة زلويغر يظهر لديهم ارتفاع بتركيز الحموض الدهنية الطويلة جدا في بلازما الدم. إن الخلايا التي تتم تنميتها في المختبر من خلايا ليمفية مأخوذة من جلد المريض تظهر ارتفاع بتركيز الحموض الدهنية الطويلة، وضعف في إنتاج أكسيد البيتا، ألفا أكسيد حمض الفيتانك، وحمض البريستان من الحموض الدهنية الطويلة.
العلاج
بسبب الخطأ في الامتصاص الناجم عن قلة الأحماض المرارية فقد تم طرح الأغذية المعطى للأطفال كعلاج بما أنها تحتوي على الأحماض الأمينية المكسرة وكمية قليلة من الدهون بحيث أن أقل من 3٪ من السعرات الحرارية قادمة من السلاسل الطويلة ثلاثية الجليسرول. ولكن فإن تقليل كمية الحموض الدهنية الطويلة لم يثبت من تقليل كمية الحموض الدهنية الطويلة في الدم [13][14]، وغالبا ما يكون سبب ذلك قدرة الإنسان على إنتاج معظمها بنفسه. إن تركيز الحموض الدهنية الطويلة يقل في بلازما الدم في حال قلة كمية الحموض الدهنية الطويلة الناتجة من الأكل بالإضافة إلى أخذ زيت لورنزو المكون من 4 اجزاء من جليسرول ثلاثي الاولييت وجزء من جليسيرول ثلاثي ايروكيت.[15] بما ان عملية تصنيع حمض الدوكساهيكسانويك تضررت [16] فإن إضافة هذا الحمض للحمية الغذائية للمريض كان اقتراحا لحل المشكلة. لكن دراسة اثبتت ان البلاسيبو اعطى نفس النتيجة.[17] ينصح باخذ الفيتامينات التي تذوب في الدهون (ا، د، ي، ك) بسبب الخلل في تصنيع الأحماض المرارية.
أثبتت دراسة جديدة علاج ناجح للعظام الضعيفة لدى المرضى باستخدام أدوية البيسفوسفونات، ولكن يجب أخذ رأي طبيب مختص في العظام قبل وصف هذه الأدوية للمريض. وقد أثبت أن تمارين الأوزان تقلل من خسارة كثافة العظام لدى الأطفال مصابين بالمرض.
توقعات سير المرض
في الوقت الحالي، لا يوجد أي علاج أو مسار علاجي ثابت ومعروف لمتلازمة زلويغر. يجب حماية المصاب من العدوى بالأمراض المخلتفة والالتهابات المختلفة لمنع مضاعفات كالتهاب الرئة والضائقة التنفسية. تعد المسارات العلاجية الأخرى فقط مسارات رعاية لتخفيف أعراض المرض. لا ينجو المرضى في العادة إلى ما بعد سن الواحدة من العمر.[18]
توجهات مستقبلية للمرض
أساليب العلاج التي تم ذكرها سابقا هي نقطة بداية للتعامل الشخصي مع المرض على أساس طبي. مع ذلك، فمن المتوقع أن تتطور هذه المعلومات إلى أساليب علاجات مستجدة يتم تجربتها في المختبرات ومع الوقت الوصول إلى مرحلة التجارب السريرية. يعطي ملف شامل لتجارب مختبرية ونماذج حيوية كاملة لمتلازمة زلويغر الأساس للتجارب المختبربة. تعد خلايا المرضى المزروعة، كخلايا الجلد الليفية، طريقة قيمة لفحص العلاجات الدوائية في المختبر. حديثا، تم إعادة برمجة الخلايا الليفية المستمدة لمرضى متلازمة زلويغر إلى خلايا جذعية محفزة تتمايز إلى نماذج خلايا عصبية وخلايا كبد تستخدم في فحوصات الأدوية. يوجد عدة نماذج فئران تم تعديل جينات PEX لديها ليكون لديها خلل في تصنيع البيروكسيسومات، بالإضافة إلى ذلك، يوجد بعض الكائنات كاللافقاريات، الديدان، ذباب الفاكهة، وبعض أنواع الأسماك التي تم تعديل جينات PEX لديها أيضا، مما أتاح مجالا لتجربة علاجات جديدة للمرض على نطاق كائن حي كامل.[4]
إن مبادئ طرق العلاج التي يتم السعي ورائها لعلاج المرض تعتمد على فحص عدة مواد كيميائية تساعد على تصنيع البيروكسيسومات وتلعب دورا في أداء وظائفها، بالإضافة إلى طرق علاج جينية وخلوية. أظهرت فحوصات السائل المنوي أن مركب البيتائين له دور كتشابيرون الذي بإمكانه أن بساعد على تجميع البيروكسيسومات في خلايا مرضى متلازمة زلويغر. وأظهرت تجارب أدوية محتملة للمرض أن مركب الأرجينين قد يعمل أيضا كتشابيرون للبيروكسيسومات. التقدم في العلاج الجيني كأنظمة نقل الجينات باستخدام الفيروس المتعلق بالغدة الكظرية (AAV) بإمكانه أن يوفر أملا في علاج أمراض وراثية كثيرة من ضمنها متلازمة زلويغر. نجاحات عدة في علاج مرض ليبر اموريسس الخلقي (LCA)، والذي هو مرض نادر يصيب العين، باستخدام نقل الجينات، قد تكون ذات أثر على علاج مرض زلويغر. بدأت تجارب نقل الجينات باستخدام فيروس AAV9 على نماذج فئران مصابة بالعمى إثر ضمور شبكية العين نتيجة متلازمة زلويغر. بما أن متلازمة زلويغر هي مرض يصيب الجسم كله، فإن العلاجات بنقل الجينات تهدف لإصلاح أعضاء مختلفة من الجسم، مع ذلك، فإن أهم عضوين في تجارب نقل الجينات لدى المرضى هما الجهاز العصبي المركزي والكبد. وأيضا، بدأت دراسات لنقل خلايا جذعية مكان الخلايا المتضررة لدى مرضى متلازمة زلويغر.[4]
مراجع
- Brul, S; Westerveld, A; Strijland, A; Wanders, R J; Schram, A W; Heymans, H S; Schutgens, R B; van den Bosch, H; Tager, J M (1988-06-01). "Genetic heterogeneity in the cerebrohepatorenal (Zellweger) syndrome and other inherited disorders with a generalized impairment of peroxisomal functions. A study using complementation analysis". Journal of Clinical Investigation (باللغة الإنجليزية). 81 (6): 1710–1715. doi:10.1172/jci113510. ISSN 0021-9738. PMID 2454948. مؤرشف من الأصل في 19 يونيو 2018. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Zellweger's syndrome at Who Named It?
- Wiedermann, H.-R. (1991-05-01). "Hans-Ulrich Zellweger". European Journal of Pediatrics (باللغة الإنجليزية). 150 (7): 451–451. doi:10.1007/BF01958418. ISSN 0340-6199. مؤرشف من الأصل في 19 يونيو 2018. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Braverman, Nancy E.; Raymond, Gerald V.; Rizzo, William B.; Moser, Ann B.; Wilkinson, Mark E.; Stone, Edwin M.; Steinberg, Steven J.; Wangler, Michael F.; Rush, Eric T. (2016-3). "Peroxisome biogenesis disorders in the Zellweger spectrum: An overview of current diagnosis, clinical manifestations, and treatment guidelines". Molecular genetics and metabolism. 117 (3): 313–321. doi:10.1016/j.ymgme.2015.12.009. ISSN 1096-7192. PMID 26750748. مؤرشف من الأصل في 17 ديسمبر 2019. الوسيط
|CitationClass=تم تجاهله (مساعدة); تحقق من التاريخ في:|تاريخ=(مساعدة) - Steinberg, Steven J.; Dodt, Gabriele; Raymond, Gerald V.; Braverman, Nancy E.; Moser, Ann B.; Moser, Hugo W. (2006-12). "Peroxisome biogenesis disorders". Biochimica et Biophysica Acta (BBA) - Molecular Cell Research. 1763 (12): 1733–1748. doi:10.1016/j.bbamcr.2006.09.010. ISSN 0167-4889. مؤرشف من الأصل في 06 ديسمبر 2019. الوسيط
|CitationClass=تم تجاهله (مساعدة); تحقق من التاريخ في:|تاريخ=(مساعدة) - GeneReviews: Peroxisome Biogenesis Disorders, Zellweger Syndrome Spectrum
- Krause, Cindy; Rosewich, Hendrik; Thanos, Melissa; Gärtner, Jutta (2006-11). "Identification of novel mutations inPEX2,PEX6,PEX10,PEX12, andPEX13in Zellweger spectrum patients". Human Mutation (باللغة الإنجليزية). 27 (11): 1157–1157. doi:10.1002/humu.9462. ISSN 1059-7794. مؤرشف من الأصل في 13 ديسمبر 2019. الوسيط
|CitationClass=تم تجاهله (مساعدة); تحقق من التاريخ في:|تاريخ=(مساعدة) - Raymond, G. V.; Watkins, P.; Steinberg, S.; Powers, J. (2009). Handbook of Neurochemistry and Molecular Neurobiology (باللغة الإنجليزية). Springer, Boston, MA. صفحات 631–670. doi:10.1007/978-0-387-30378-9_26. ISBN 9780387303451. مؤرشف من الأصل في 22 يونيو 2018. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Woliver, R. (2009). Alphabet Kids - From ADD to Zellweger Syndrome. London, UK: Jessica Kingsley.
- Online Mendelian Inheritance in Man (OMIM) Zellweger syndrome; ZS -214100
- Steinberg, S.; Chen, L.; Wei, L.; Moser, A.; Moser, H.; Cutting, G.; Braverman, N. (2004). "The PEX Gene Screen: molecular diagnosis of peroxisome biogenesis disorders in the Zellweger syndrome spectrum". Molecular Genetics and Metabolism. 83 (3): 252–263. doi:10.1016/j.ymgme.2004.08.008. PMID 15542397.
- Yik, W. Y.; Steinberg, S. J.; Moser, A. B.; Moser, H. W.; Hacia, J. G. (2009). "Identification of novel mutations and sequence variation in the Zellweger syndrome spectrum of peroxisome biogenesis disorders". Human Mutation. 30 (3): E467–E480. doi:10.1002/humu.20932. PMC 2649967 . PMID 19105186.
- Van Duyn, MA; Moser, AE; Brown FR, 3rd; et al. (August 1984). "The design of a diet restricted in saturated very long-chain fatty acids: therapeutic application in adrenoleukodystrophy". The American Journal of Clinical Nutrition. 40 (2): 277–84. doi:10.1093/ajcn/40.2.277. PMID 6465061.
- Brown FR, 3rd; Van Duyn, MA; Moser, AB; et al. (October 1982). "Adrenoleukodystrophy: effects of dietary restriction of very long chain fatty acids and of administration of carnitine and clofibrate on clinical status and plasma fatty acids". The Johns Hopkins medical journal. 151 (4): 164–72. PMID 7120720.
- Moser, AB; Borel, J; Odone, A; et al. (March 1987). "A new dietary therapy for adrenoleukodystrophy: biochemical and preliminary clinical results in 36 patients". Annals of Neurology. 21 (3): 240–9. doi:10.1002/ana.410210305. PMID 2440378.
- Martinez, M (26 June 1992). "Abnormal profiles of polyunsaturated fatty acids in the brain, liver, kidney and retina of patients with peroxisomal disorders". Brain Research. 583 (1–2): 171–82. doi:10.1016/s0006-8993(10)80021-6. PMID 1504825.
- Paker, AM; Sunness, JS; Brereton, NH; et al. (31 August 2010). "Docosahexaenoic acid therapy in peroxisomal diseases: results of a double-blind, randomized trial". Neurology. 75 (9): 826–30. doi:10.1212/WNL.0b013e3181f07061. PMC 3013498 . PMID 20805528.
- Steinberg, S.; Dodt, G.; Raymond, G.; Braverman, N.; Moser, A.; Moser, H. (2006). "Peroxisome biogenesis disorders". Biochimica et Biophysica Acta (BBA) - Molecular Cell Research. 1763 (12): 1733–48. doi:10.1016/j.bbamcr.2006.09.010. PMID 17055079.
وصلات خارجية
- Zellweger syndrome at Zellweger UK
- Zellweger-Syndrome على موقع المعهد الوطني للاضطرابات العصبية والسكتة الدماغية (NINDS)
- Health Link at كلية طب ويسكونسن
- The Global Foundation for Peroxisomal Disorders
 بوابة طب
بوابة طب
