متلازمة دي جورج
متلازمة حذف 22q11.2، تعرف أيضا بمتلازمة دي جورج، شذوذ دي جورج، متلازمة الحنك والقلب والوجه، متلازمة شبرينتزين، متلازمة سترونج، عدم تنسج الصعترية الخلقي، ونقص نمو الغدة الصعترية ، وهي متلازمة تحدث نتيجة حذف جزء صغير من الصبغي أو (الكروموسوم) رقم 22 الحذف يحدث بالقرب من منتصف كروموسوم في مكان معين q11.2 أي على ذراع طويلة من واحد من زوج من الكروموسومات 22. لديها انتشار تقدر 1:4000.[2] وقد وصفت المتلازمة في عام 1968 من قبل طبيب غدد الأطفال أنجيلو دي جورج.
| متلازمة دي جورج | |
|---|---|
 تخفيضات التصوير المقطعي بالكمبيوتر للمريض ، مما يدل على العقد القاعدية والتكلس حول البطين. من تقرير حالة بقلم تونيلي وآخرون ، 2007[1] تخفيضات التصوير المقطعي بالكمبيوتر للمريض ، مما يدل على العقد القاعدية والتكلس حول البطين. من تقرير حالة بقلم تونيلي وآخرون ، 2007[1] | |
| معلومات عامة | |
| الاختصاص | علم الوراثة الطبية |
| من أنواع | متلازمة ، ومرض وراثي سائد |
وصف الحالة
ملامح هذه المتلازمة تختلف على نطاق واسع، حتى بين أفراد من عائلة واحدة، وتؤثر على أجزاء كثيرة من الجسم. العلامات المميزة والأعراض قد تشمل العيوب الخلقية مثل أمراض القلب الخلقية(congenital heart diseas) وعيوب في الحنك (palate defect)غالبا ما تتعلق بمشاكل عصبية-عضلية في الإغلاق (ضعف الحنك والبلعوم),صعوبات التعلم، اختلافات طفيفة في ملامح الوجه، والتهابات أو عدوى متكررة. العدوى شائعة في الأطفال بسبب مشاكل في المناعة المرتبطة بالخلايا تي في الجهاز المناعي، بسبب غياب أو ضمور الغدة الصعترية.متلازمة حذف 22q11.2 قد تلاحظ لأول مرة عندما يظهر المولود المصاب عيوبا في القلب أو تشنجات نتيجة لنقص الكالسيوم في الدم، بسبب خلل في وظائف الغدد الجاردرقية ونقص مستويات هرمون الباراثورمون. الأفراد المصابين قد يكون لديهم أيضا أي نوع آخر من العيوب الخلقية، بما في ذلك عيوب الكلية الخلقية، وصعوبات ملحوظة في التغذية كأطفال. أمراض المناعة الذاتية، مثل نقص افراز الغدة الدرقية، أو نقص افراز الغدة الجاردرقية أو نقص الصفائح الدموية في الدم، والأمراض النفسية هي اعراض أخرى شائعة الظهور في المراحل المتأخرة من المرض الحذف المصغر في المنطقة الصبغية 22q11.2 مرتبطة بزيادة 20-30 ضعفا في خطر الإصابة بانفصام الشخصية.
الدراسات تقدم معدلات متفاوتة من الحذف في المنطقة الصبغية 22q11.2 في حالات انفصام الشخصية، تتراوح من 0.5 إلى 2% ,بمتوسط 1% مقارنة بالنسبة الكلية المقدرة لحدوث متلازمة حذف 22q11.2 في المجتمع السكاني وهي 0.025%.
يمكن للطلاب الطب استخدام ذاكري CATCH-22 لوصف متلازمة دي جورج، مع استخدام 22 لتذكيرهم بأن الخلل يحدث في الصبغي رقم 22 كما يلي :[3]
Cardiac Abnormality (especially tetralogy of Fallot)
وتعني حدوث عيوب في القلب، خاصة رباعية فالو
Abnormal facies
ملامح غير طبيعية
Thymic aplasia
لا تنسج الغدة الصعترية
Cleft palate
الحنك المشقوق
Hypocalcemia.
نقص نسبة الكالسيوم في الدم
المصطلحات
علامات وأعراض متلازمة حذف 22q11 متنوعة بحيث كان ينظر للمجموعات المختلفة من الأعراض على أنها حالات منفصلة، وشملت هذه التصنيفات الأصلية متلازمة الحنك والقلب والوجه، متلازمة دي جورج ,متلازمة شبرينتزين ,متلازمة سيدلاكوفا ,ومتلازمة شذوذ الوجه المخروطي، ومن المفهوم الآن أن تكون جميع هذه العروض من متلازمة واحدة.
الأعراض
لأفراد المصابين بحذف 22q11.2 يمكن أن يعانوا من العديد من المظاهر المحتملة، تتراوح في عدد من المزايا المرتبطة بها ومن خفيفة إلى خطيرة جدا. تبين أن الأعراض شيوعا تشمل :
- أمراض القلب الخلقية (40 ٪ من الأفراد)، خاصة (رباعية فالوت، قوس الأبهرالمنقطع، عيب الحاجز البطيني، وبقاء الجذع الشرياني مفتوحا
- عيوب الحنك (50%), خصوصا عدم توافق الحنك والبلعوم، وشق الحنك تحت المخاطي، والحنك المشقوق، وخصائص مميزة في الوجه في أغلبية الأفراد القوقازيين مثل فرط التباعد
- صعوبات التعلم (90 ٪)، ولكن مجموعة واسعة
- hypocalcemia (50 ٪) (نتيجة لنقص نشاط الغدد الجاردرقية)
- مشاكل كبيرة في التغذية (30 ٪)
- العيوب الخلقية في الكلى (37 ٪)
- فقدان السمع (سواء موصل والحسي العصبي) (فقدان السمع مع متلأزمات الجمجمة)
- عيوب في الحنجرة والقصبة الهوائية والمريء
- نقص هرمون النمو
- اضطرابات المناعة الذاتية
- نوبات من التشنجات (بدون انخفاض مستوى الكالسيوم في الدم
- عيوب الهيكل العظمي
الأسباب
المرض يحدث عن طريق الحذف الوراثي (فقدان جزء صغير من المادة الوراثية)الواقع على ذراع طويلة من واحد من اثنين من الكروموسومات 22. نادرا جدا، مرضى بمظاهر سريرية مشابهة قد يعانون من حذف للمادة الوراثية على الذراع القصيرة من الصبغي رقم 10
الآلية التي تسبب كل من الميزات المرتبطة بها من متلازمة غير معروفة. متلازمة حذف 22q11.2 قد تتضمن عيوبا في هجرة الأنسجة المنشقة من الحد العصبي، تؤثر بشكل خاص في نمو الجيوب البلعومية رقم 3 و4. وهذا يؤثر على الغدة الصعترية ؛عضو في منطقة الصدر مسئول بنسبة كبيرة عن تمايز واستحثاث التسماح في الخلايا المناعية تي, كذلك يؤثر على الغدد الجاردرقية، المسئولة عن تنظيم مستويات الكالسيوم في الدم
العلاج
لا يوجد علاج جيني لمتلازمة حذف 22q11.2. ميزات فردية معينة يمكن علاجها باستخدام العلاجات القياسية والمعروفة. طريقة العلاج تعتمد على تحديد كل الأعراض المترابطة، ومعالجة كل منها باستخدام أفضل السبل المتاحة
على سبيل المثال، لدى الأطفال من المهم أن يتم تحديد مشاكل جهاز المناعة في وقت مبكر ما يلزم اتخاذ احتياطات خاصة فيما يتعلق بنقل الدم وإعطاء لقاحات حية. ذراعة الغدة الصعترية قد يستخدم لعلاج غياب هذه الغدة في الحالات النادرة والمعروفة بتناذر دي جورج الكامل. علاج الالتهابات البكتيرية هي بالمضادات الحيوية. قد يتطلب العلاج أيضا عمليات جراحية في القلب لتصحيح العيوب الخلقية. نقص نشاط جارات الدرق يسبب نقص الكالسيوم في الدم وغالبا ما يتطلب استخدام فيتامين (د) مدى الحياة ومكملات الكالسيوم.
التشخيص والاختبارات
يتم تشخيص متلازمة حذف 22q11.2 في الأشخاص الذين يعانون من الحذف تحت المجهري من كروموسوم 22 بواسطة وميض التهجين الموضعي باستخدام مجس الحمض النووي من الجزء 22q11.2 الصبغي. مثل هذا الاختبار الجيني متوافر على نطاق واسع للاختبار السريريو فترة ما قبل الولادة لمتلازمة حذف 22q11.2. أقل من 5 ٪ من الأفراد مع الأعراض السريرية لتناذر حذف 22q11.2 لديهم اختبارات خلوية اعتيادية، واختبار وميض تهجين موضعي سلبي. قد يكون لديهم محذوفات معينة من تناذر دي جورج قابلة للتكشف فقط بالطرق الاختبارية أو باستعمال وسائل اختبارات سريرية متقدمة أكثر.
الوراثيات
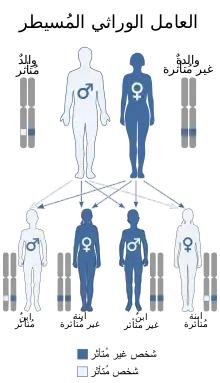
معظم الناس مع متلازمة حذف 22q11.2 يفقدون حوالي 3 ملايين زوج قاعدة ثانية (وحدات بناء الحمض النووي) على نسخة واحدة من كروموسوم 22 في كل خلية من خلايا الجسم. هذه المنطقة تحتوي على حوالي 45 من الجينات، ولكن بعض هذه الجينات لم يتم وصفها بشكل دقيق. وهناك نسبة صغيرة من الأفراد المتضررين لديهم حذوفات أصغر في نفس المكان.
الباحثون لم يتعرفوا بعد على الجينات التي تسهم في ملامح متلازمة حذف 22q11.2. تحدد لديهم أن فقدان جين واحد بشكل خاص على الصبغي 22، TBX1، هو على الارجح مسؤولة عن بعض من متلازمة المميز للعلامات (مثل عيوب القلب). حمل نسخة واحدة فقط من هذا الجين لا يبدو أن سبب صعوبات التعلم، ولكن. جينات أخرى في المنطقة المحذوفة من المرجح أن تسهم في علامات وأعراض متلازمة حذف 22q11.2 والجينات خارج المنطقة 22q11.2 قد تلعب أيضا دورا.
قد يكون تناذر حذف 22q11.2 وراثيا، ولكن هذا هو حال الأقلية من الأشخاص المشخصون بهذا المرض مؤخرا 5-10 ٪ فقط قد ورثت حذف 22q11.2 من أحد الوالدين، في حين أن نحو 90-95 ٪ من الحالات يكون لها حذف جديد (جديد للأسرة) للمنطقة 22q11.2. وذلك لأن المنطقة 22q11.2 لديها البنية التي تجعلها عرضة لإعادة تنظيم خلال تكوين الحيوان المنوي أو البويضة الحذف قابل للحدوث بصورة متساوية تقريبا في حالة تكوين البويضة لحالة تكوين الحيوان المنوي فرد مع حذف 22q11.2 لديه 50 ٪ (واحد في اثنين) فرصة لتمرير حذف 22q11.2 لذريتهم. اختبار ما قبل الولادة، مثل بزل السلى، يتوفر لحالات الحمل التي يتقرر انها في خطر الإصابة بهذا المرض. أيضا، والحمل مع إيجاد أمراض القلب الخلقية و/ أو شذوذ الحنك المكتشفة بواسطة الفحص بالموجات فوق الصوتية يمكن أن تخضع لاختبارات ما قبل الولادة لمتلازمة حذف 22q11.2. لأن علامات هذه المجموعة من العيوب يمكن أيضا أن يكون معظم الموروثة وراثي جسميأو مرتبط بالجين X أو الصفات المتنحية فقطاختبار وراثي من كلا الأبوين يمكن أن يحدد على وجه اليقين احتمالية حدوث أي من هذه الحالات الشاذة في الأطفال اللاحقين. لا استشهادات
الانتشار
متلازمة حذف 22q11.2 يؤثر على ما يقدر ب 1 من كل 4000 ولادة حية.[2] ويستند هذا التقدير على العيوب الخلقية الكبرى وقد يكون أقل من الواقع، لأن العديد من الأفراد الماصبين بالحذف لديهم أعراضا قليلة وقد لا يكونوا تم تشخيصهم من قبل
مشاكل إدراكية ولغوية
الضعف الإدراكي
الأطفال الذين يعانون من حذف 22q11.2 لديهم بيانات محددة في الاختبارات النفسية العصبية. وعادة يكون لهم معدل ذكاء على الحد الفاصل، وغالبية الأفراد يحصلون على درجات أعلى في الاختبارات اللفظية دونا عن غيرها من الاختبارات الأداء الإدراكي عند معالجة المعلومات التي تنطوي على المكان والزمان عادة ما يظهر ضعف كبير وهذا يؤدي إلى إبطاء عام في تطوير المعارف والمهارات العددية والحسابية.
والجدير ذكره ان هؤلاء المرضى هم مجموعة خاصة ذات الخطورة العالية لتطوير الفصام. 30 ٪ على الأقل لديهم حادثة واحدة من الذهان و25 % يصابون بمرض الفصام فعلا.[4]
الكلام واللغة
الأبحاث الحالية تثبت وجود بيانات فريدة عن الكلام وضعف في اللغة مرتبط بتناذر حذف 22q11.2. في اختبارت تقييم معدل الذكاء، يؤدي الأطفال نتائج أقل في التخاطب واللغة مقارنة بنتائج الاختبارات غير اللفظية تتضمن المشاكل الشائعة الخنة المفرطة، وتأخر اللغة، وأخطاء مخارج الحروف.[5][6][7]
فرط الخنة يحدث عندما يهرب الهواء من خلال الأنف أثناء إنتاج أصوات الكلام الشفوي مما يؤدى إلى انخفاض وضوح الكلام. هذا هو سمة مشتركة في التعبير والتعريف باللغة لأن 69 ٪ من الأطفال يعانون من التشوهات الحنكية. وإذا كان هيكل غشاء اللهاة هو من النوع الذي لا يوقف تدفق الهواء من الصعود إلى تجويف الأنف، فإن ذلك سيكون سببا لاختنان الصوت ويشار إلى هذه الظاهرة بأنها عدم كفاءة الحنك والبلعوم. فقدان السمع يمكن أن تسهم أيضا في زيادة فرط الخنة لأن الأطفال ضعاف السمع يمكن أن يجدوا صعوبة في مراقبة الكلام الصادر منهم. خيارات العلاج المتاحة لنقص كفاءة اللهاة والبلعوم تتضمن التعويض والجراحة.[5][8][9][10]
صعوبات الحصول على المفردات وصياغة اللغة المحكية (عجز اللغة التعبيرية) في بداية تطوير اللغة هي أيضا جزء من لغة الكلام والتعريف المرتبطة بحذف 22q11.2. وغالبا ما يتأخر اكتساب المفردات بشدة في عمر ما قبل المدارس في هؤلاء الأطفال في بعض الدراسات الحديثة، كان للأطفال مجموعة مفردات محدودة للغاية، أو كانوا بدون كلام في سن سنتين إلى ثلاثة من العمر الأطفال في سن المدرسة يحرزون تقدما في لغتهم التعبيرية بينما يكبرون، لكن العديد منهم يظهرون تأخيرا وصعوبة في تذكر بعض الكلمات وإنتاج جمل أطول وأكثر تعقيدا لغة المتقبل، وهي القدرة على فهم، أو الحتفاظ، أو معالجة اللغة المنطوقة قد تشمل صعوبات أيضا، ولكن عادة ليست بنفس شدة الصعوبات كاللغة التعبيرية
أخطاء النطق أيضا شائعة في الأطفال المصابين بتناذر حذف 22q11.2. هذه الأخطاء تشمل أيضا مخارج حروف محدودة، مما يضطر الطفل لاستخدام طرق نطق تعويضية بديلة مما يقلل التفاهم المتبادل للطفل وقدرته على التواصل. مخارج الحروف عادة تتكون من الأصوات المحدثة في مقدمة القناة الصوتية أو مؤخرتها مثل \ب\,\و\,\ج\,\م\,\ن\ وفواصل لسان المزمار. أصوات منتصف القناة الصوتية غائبة تماما. الأخطاء التعويضية التي يرتكبها هؤلاء الأطفال تشمل:فواصل لسان المزمار، بدائل أنفية، حروف احتكاكية بلعومية، حروف الصفير الشفوية، تقليل الضغط على مشيرات السواكن، أو مجموعة من هذه الأعراض. الأكثر شيوعا بين هذه الأخطاء هي فواصل لسان المزمار (نطق الهمزات). تحدث هذه الأخطاء السابقة، والمخارج البديلة نتيجة العيوب التكوينية في الحنك. صعوبات النطق التي تظهرها هذه الفئة تكون أكثر حدة في السن الصغير، وتظهر تحسنا تدريجيا أثناء النمو والنضوج
المراجع
- Tonelli AR, Kosuri K, Wei S, Chick D (2007). "Seizures as the first manifestation of chromosome 22q11.2 deletion syndrome in a 40-year old man: a case report". J Med Case Reports. 1: 167. doi:10.1186/1752-1947-1-167. PMC 2222674. PMID 18053182. الوسيط
|CitationClass=تم تجاهله (مساعدة)صيانة CS1: أسماء متعددة: قائمة المؤلفون (link) - Oskarsdóttir S, Vujic M, Fasth A (2004). "Incidence and prevalence of the 22q11 deletion syndrome: a population-based study in Western Sweden". Arch. Dis. Child. 89 (2): 148–51. doi:10.1136/adc.2003.026880. PMC 1719787. PMID 14736631. الوسيط
|CitationClass=تم تجاهله (مساعدة)صيانة CS1: أسماء متعددة: قائمة المؤلفون (link) - Burn, J (1999). "Closing time for CATCH22". J. Med. Genetics. 36: 737–38. doi:10.1136 (غير نشط 2009-11-14) تأكد من صحة قيمة
|doi=(مساعدة). الوسيط|CitationClass=تم تجاهله (مساعدة) - Zinkstok J, van Amelsvoort T (2005). "Neuropsychological profile and neuroimaging in patients with 22Q11.2 Deletion Syndrome: a review". Child Neuropsychol. 11 (1): 21–37. doi:10.1080/09297040590911194. PMID 15823981. الوسيط
|CitationClass=تم تجاهله (مساعدة) - D'Antonio LL, Scherer NJ, Miller LL, Kalbfleisch JH, Bartley JA (2001). "Analysis of speech characteristics in children with velocardiofacial syndrome (VCFS) and children with phenotypic overlap without VCFS". Cleft Palate Craniofac. J. 38 (5): 455–67. doi:10.1597/1545-1569(2001)038<0455:AOSCIC>2.0.CO;2. PMID 11522167. الوسيط
|CitationClass=تم تجاهله (مساعدة)صيانة CS1: أسماء متعددة: قائمة المؤلفون (link) - Scherer NJ, D'Antonio LL, Kalbfleisch JH (1999). "Early speech and language development in children with velocardiofacial syndrome". Am. J. Med. Genet. 88 (6): 714–23. doi:10.1002/(SICI)1096-8628(19991215)88:6 (غير نشط 2009-11-14). PMID 10581495. الوسيط
|CitationClass=تم تجاهله (مساعدة)صيانة CS1: أسماء متعددة: قائمة المؤلفون (link) - Scherer NJ, D'Antonio LL, Rodgers JR (2001). "Profiles of communication disorder in children with velocardiofacial syndrome: comparison to children with Down syndrome". Genet. Med. 3 (1): 72–8. PMID 11339384. الوسيط
|CitationClass=تم تجاهله (مساعدة)صيانة CS1: أسماء متعددة: قائمة المؤلفون (link) - Eliez S, Palacio-Espasa F, Spira A (2000). "Young children with Velo-Cardio-Facial syndrome (CATCH-22). Psychological and language phenotypes". Eur Child Adolesc Psychiatry. 9 (2): 109–14. doi:10.1007/s007870050005. PMID 10926060. الوسيط
|CitationClass=تم تجاهله (مساعدة)صيانة CS1: أسماء متعددة: قائمة المؤلفون (link) - Robin NH, Shprintzen RJ (2005). "Defining the clinical spectrum of deletion 22q11.2". J. Pediatr. 147 (1): 90–6. doi:10.1016/j.jpeds.2005.03.007. PMID 16027702. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Solot CB, Knightly C, Handler SD (2000). "Communication disorders in the 22Q11.2 microdeletion syndrome". J Commun Disord. 33 (3): 187–203, quiz 203–4. doi:10.1016/S0021-9924(00)00018-6. PMID 10907715. الوسيط
|CitationClass=تم تجاهله (مساعدة)صيانة CS1: أسماء متعددة: قائمة المؤلفون (link)
هذا المقال يتضمن النص المجال العام من المكتبة الوطنية الأمريكية للطب
وصلات خارجية
- متلازمة دي جورج على مشروع الدليل المفتوح
- Velo - القلب والوجه مؤسسة متلازمة المعلومات
- GeneReviews / نكبي / المعاهد الوطنية للصحة دخول اتحاد الحرية / متلازمة نقص في 22q11.2 الحذف
- GeneReviews / نكبي / المعاهد الوطنية للصحة دخول اتحاد الحرية / متلازمة الازدواجية في 22q11.2
- معلومات عن متلازمة الحذف 22q11.2 من مستشفى سياتل للأطفال
 بوابة علم الأحياء الخلوي والجزيئي
بوابة علم الأحياء الخلوي والجزيئي بوابة طب
بوابة طب
