متلازمة القرن القذالي
متلازمة القرن القذالي، كانت تعرف سابقًا باسم متلازمة إهلرز-دانلوس,[1] هو اضطراب في النسيج الضام ( مرض وراثي متنحي مرتبط X ). وهو ناتج عن نقص النحاس ( من المعادن الأساسية في الجسم )، ويرتبط ذلك المرض ب طفرة في جين ATP7A.[2][3] حوالي 2/3 من الأطفال الذين يعانون من متلازمة القرن القذالي بسبب تلك الطفرة الوراثية. بينما 1/3 الأطفال الآخرين لم يكن المرض في اسرهم ( أي ليس مرض وراثي ).هذه المتلازمة تصيب الذكور بشكل أكبر وذلك لاحتواء الجينات الذكرية على جين X واحد على عكس الإناث التي تكون في الأغلب حاملة للمرض فقط ولكى تصاب الإناث لابد من أن يكون المرض على ال 2 كروموسوم X فيما عدا ذلك تكون حاملة للمرض فقط[4] هذا الاضطراب يعتبر أقل من مرض مينكيس.[5]
| متلازمة القرن القذالي | |
|---|---|
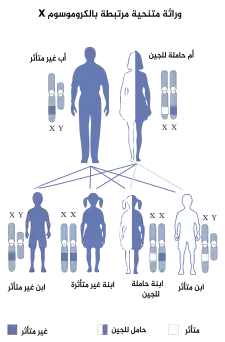 وراثة متلازمة القرن القذالي وراثة متلازمة القرن القذالي | |
| معلومات عامة | |
| الاختصاص | علم الغدد الصم |
علامات / أعراض
هذه المتلازمة تتميز بنقص في صفراء نحاس مما يؤدي إلى تشوهات في الهيكل العظمي. ويشمل الجزء الخلفي من الجمجمة وكذلك تشوهات في الكوع و المرفق, و رأس الكعبرة و تشوهات الوركين والحوض.[1] متلازمة القرن القذالي تأتي في وقت مبكر في الطفولة الوسطى.[4] ومن بعض هذه السمات في الأطفال :
- طبيعي / تأخر قليل بمعدل الذكاء
- رقبة طويلة، وارتفاع سقف الحلق -، الوجه طويل، والجبهه عالية
- رخاوة الجلد
- الفتوق الأربية
- التواء الأوعية الدموية
- رتوج المثانة
- خلل الوظائف المستقلة—عدم القدرة على تنظيم أجزاء من الجهاز العصبي
- الإسهال المزمن
- شعر خشن
العلاج
الأطفال الذين يعانون من متلازمة القرن القذالي يحصلون على العلاج الطبيعي والمهني.[4] قد يتطلب أنبوب تغذية إذا كانوا لا ينمون بما فيه الكفاية. في محاولة لتحسين الحالة العصبية (التشنجات) حقن كلوريد النحاس يمكن أن يُعطى للطفل في وقت مبكر. ومع ذلك، فقد أظهرت حقن النحاس انها غير فعالة في دراسات اضطرابات الأنسجة الضامة الأيضية.[6]
مراجع
- الوراثة المندلية البشرية عبر الإنترنت (OMIM) 304150
- Scheiber, Ivo; Dringen, Ralf; Mercer, Julian F. B. (2013). "Chapter 11. Copper: Effects of Deficiency and Overload". In Astrid Sigel, Helmut Sigel and Roland K. O. Sigel (المحرر). Interrelations between Essential Metal Ions and Human Diseases. 13. Springer. صفحات 359–387. doi:10.1007/978-94-007-7500-8_11. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Tang J, Robertson S, Lem KE, Godwin SC, Kaler SG (نوفمبر 2006). "Functional copper transport explains neurologic sparing in occipital horn syndrome". Genet. Med. 8 (11): 711–8. doi:10.1097/01.gim.0000245578.94312.1e. PMID 17108763. مؤرشف من الأصل في 28 مايو 2012. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Horn Syndrome, 9 August 2004. [وصلة مكسورة] نسخة محفوظة 11 أكتوبر 2016 على موقع واي باك مشين. [وصلة مكسورة] نسخة محفوظة 11 أكتوبر 2016 على موقع واي باك مشين.
- Kennerson ML, Nicholson GA, Kaler SG, et al. (مارس 2010). "Missense mutations in the copper transporter gene ATP7A cause X-linked distal hereditary motor neuropathy". Am. J. Hum. Genet. 86 (3): 343–52. doi:10.1016/j.ajhg.2010.01.027. PMC 2833394. PMID 20170900. مؤرشف من الأصل في 13 ديسمبر 2019. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Kodama, H.; C. Fujisima, W. Bhadhprasit. (نوفمبر 2010). "Pathology, clinical features and treatments of congenital copper metabolic disorders - Focus on neurologic aspects". Brain & Development. الوسيط
|CitationClass=تم تجاهله (مساعدة)
وصلات خارجية
- GeneReviews/NCBI/NIH/UW entry on ATP7A-Related Copper Transport Disorders
- Occipital horn syndrome في معاهد الصحة الوطنية الأمريكية للأمراض النادرة
 بوابة طب
بوابة طب