متلازمة آدامز أوليفر
متلازمة آدامز أوليفر هي متلازمة نادرة تظهر في المواليد، وسماتها الأساسية هي عدم تنسج الجلد الخلقي في فروة الرأس وعيوب في الأطراف، وقد تظهر بعض السمات الأخرى في المصاب.[1]
| متلازمة آدامز أوليفر | |
|---|---|
| معلومات عامة | |
| الاختصاص | طب الأطفال ، وطب الجلد ، وجراحة العظام |
| من أنواع | متلازمة |
الانتشار

نادرة جداً ومعدل انتشارها غير معروف،[1] لكنها تصيب الذكور والإناث بنسب متساوية، وتوجد أكثر من 125 حالة مسجلة.[2]
السمات
- غياب كامل أو جزئي لطرف ما
- ارتفاق الأصابع
- كثرة الأصابع
- فرط نمو الأطراف
- عدم تنسج الجلد الخلقي في فروة الرأس، وفي الحالات الشديدة قد يختفي جزء من عظم الجمجمة أيضاً فتظهر الأنسجة تحتها.
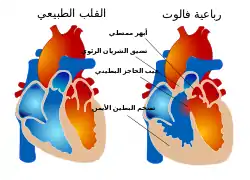
وتعاني 20% من الحالات من عيوب في القلب، مثل:
- عيب الحاجز البطيني
- عيب الحاجز الأذيني
- رباعية فالوت
- بطين أيمن ذو مخرجين
سمات إضافية تم الإبلاغ عنها:
- الجلد المرمري متوسع الشعيرات الخلقي
- ارتفاع الضغط في الشريان الرئوي
- عيوب في الجهاز العصبي المركزي[2]
الأسباب
غير معروفة، لكن توجد نظريات عديدة تُرجعها إلى عيوب في الأوعية الدموية أثناء تكوين الجنين.
وقد وصفت من قبل كوراثة صبغية جسدية سائدة؛ نظراً لإصابة أجيال متعددة في نفس العائلة، ويرجع الاختلاف في درجة الإصابة إلى اختلاف درجة التعبُّر والانتفاذ الوراثي للصبغي، وفيما بعد وجد أن بعض الأفراد أصيبوا عن طريق الوراثة الجسدية المتنحية[3]، وقد عرفت أربعة جينات للمتلازمة، هي: ARHGAP31 [الإنجليزية], Dock6, RBPJ, Notch1 [الإنجليزية][4][5]
التشخيص
عن طريق الفحص الإكلينيكي الشامل، تظهر سمات المتلازمة الرئيسية والثانوية[6]، وهي كالآتي:
| السمات الرئيسية | السمات الثانوية |
|---|---|
| عيوب الأطراف | توسع الشعيرات الدموية المرمري |
| عدم تنسج الجلد الخلقي | عيب خلقي في القلب |
| وجود سوابق عائلية للإصابة بالمتلازمة | عيب في الأوعية الدموية |
ويتم التشخيص بسمتين رئيسيتين، ووجود سمة رئيسية مع سمة ثانوية يرجح الإصابة، وحتى الآن لا يوجد اختبارات للجينات تؤكد أو تستبعد الإصابة بالمتلازمة.[3]
المضاعفات
- نزيف من الرأس أو عدوى تؤدي إلى التهاب السحايا
- إعاقة بسبب عيوب الأطراف
- العيوب الخلقية في القلب قد تؤدي إلى قصور القلب إذا لم يتم علاجها[7]
العلاج
معالجة عرضية إذ لا يمكن علاج السبب، وتعتمد على شدة الأعراض؛ فعيوب الجلد قد تترك لتختفي تلقائياً، بينما تحتاج بعض العيوب إلى علاج جراحي كمشكلات القلب والأوعية الدموية.[3]
التاريخ
تم الإبلاغ عنها لأول مرة في عائلة بها ثمانية أفراد مصابة.[8]
انظر أيضًا
المراجع
- Adams-Oliver syndrome - Genetics Home Reference نسخة محفوظة 22 ديسمبر 2017 على موقع واي باك مشين.
- Adams Oliver Syndrome - NORD (National Organization for Rare Disorders) نسخة محفوظة 20 فبراير 2017 على موقع واي باك مشين.
- Adams-Oliver syndrome | Contact a Family نسخة محفوظة 24 يناير 2017 على موقع واي باك مشين.
- Stittrich AB, Lehman A, Bodian DL, Ashworth J, Zong Z, Li H, Lam P, Khromykh A, Iyer RK, Vockley JG, Baveja R, Silva ES, Dixon J, Leon EL, Solomon BD8, Glusman G1, Niederhuber JE9, Roach JC1, Patel MS (2014) Mutations in NOTCH1 cause Adams-Oliver Syndrome. Am J Hum Genet pii: S0002-9297(14)00320-6
- OMIM Entry - # 100300 - ADAMS-OLIVER SYNDROME 1; AOS1 نسخة محفوظة 09 يوليو 2017 على موقع واي باك مشين.
- Snape et al. 2009
- Adams Oliver Syndrome نسخة محفوظة 08 أبريل 2017 على موقع واي باك مشين.
- Adams and Oliver, 1945.
