فرط تيروزين الدم I
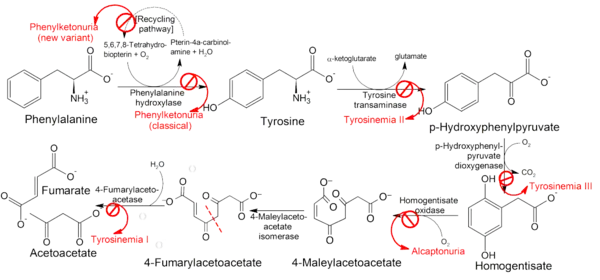
فرط تيروزين الدم I، ويعرف أيضاً بفرط تيروزين الدم الكبدي الكلوي، هو أشد أنواع فرط تيروزين الدم، وينشأ نتيجة نقص في إنزيم فوماريلاسيتوسيتات هدرولاز (fumarylacetoacetase FAH)، وهذا الإنزيم يحفز الخطوة الأخيرة من استقلاب التيروزين والفينيل الانين مما يؤدي إلى تراكم كميات كبيرة من الحمض الاميني التيروزين في الدم والأنسجة. يتميز هذا المرض باضطراب متفاقم للكبد مما يؤدي إلى خلل في الانابيب الكلوية يسبب نقص الفسفور في الدم والتالي إلى الكساح (يسمى الكساح الفسفوري). بداية ظهور أعراض المرض تختلف من شخص لآخر، بعضهم يظهر في مرحلة الطفولة والبعض في مرحلة المراهقة. في أسوأ صور المرض قد يعاني المصاب من فشل الكبد الحاد في غضون أسابيع بعد الولادة، بينما في أخف صوره قد يعاني المريض من الكساح بشكل رئيسي في التيروزينميا المزمن. المرضى الذين لم يعالجوا، يموتون من تليف الكبد أو سرطان الكبد في سن مبكرة.
| فرط تيروزين الدم I | |
|---|---|
| معلومات عامة | |
| الاختصاص | علم الغدد الصم |
| من أنواع | فرط تيروزين الدم |
طرق التوريث (الوراثيات: Genetics)
معدل الإصابة العالمي لهذا المرض الوراثي النادر هو حوالي 1 في 100,000 شخص. تعتبر النرويج من أكثر الدول تأثرا حيث يصيب حوالي 1 من كل 60,000 إلى 74,000 فرد.[1] تعتبر منطقة الكيبيك في كندا هي الأكثر اصابة عالميا حيث يصاب حوالي 1 من كل 16,000 فرد وتحديدا في منطقة ساغيناي لاك سانت جان كيبيك حيث يعتبر فرد من كل 1846 مصابا بهذا المرض.[2]
الفِيزْيُولُوجْيا المَرَضِيَّة
يعتبر فقدان كفاءة إنزيم هيدروليز فيوميريل استيت والذي يحفز الخطوة الاخيرة من عملية تحلل التيروزين هو المسبب الرئيسي لهذا المرض. يقوم هذا الإنزيم تحويل الفيوميريل استيت إلى فيومارات واستيت وسكسنيت. يتراكم الفيوميريل استيت في الكبد وانابيب الكلى ويسبب اضرار اكسدة للخلايا وحمضها النووي، مما يؤدى إلى تلف الخلايا أو اضرار بوظائفها أو وظائف جيناتها. من هذه الاضرار عرقلة بناء البروتين وعرقلة عملية استحداث الجلوكوز في الكبد وعرقلة وظائف الخلايا الموجودة في الانابيب القريبة للكلى مما يؤدي إلى خلل عام في وظائف الامتصاص والوظائف الاخرى لهذه الانابيب مسببا ما يعرف ب متلازمة فانكوني.[3] أظهر Hostetter وغيره (1983)، أن تلف الكبد بدأت في مرحلة ما قبل الولادة وأن فرط تيروزين الدم تظهر فقط بعد الولادة.[4]
الأعراض والعلامات (الصورة السريرية)
عادة ما يكون الوضع الطبيعي عند الولادة، والأطفال الذين يعانون من هذا المرض غالبا ما تتطور الأعراض خلال الأشهر القليلة الأولى من الحياة. هذه الأعراض يمكن أن تختلف اختلافا كبيرا. في كثير من الأحيان، يفشل الطفل في اكتساب الوزن وعادة ما يكون منزعجا. وتتضمن الأعراض المبكرة الأخرى الحمى والإسهال والقيء وتضخم الكبد (حتى يبدو بطن الطفل منتفخا)، كما تظهر الكدمات (وهي بقع خضراء أو بنية على الجلد)، واليرقان (حيث يبدو الجلد والعين أصفر اللون)، وقد يحدث نزيف في الأنف. إذا تركت دون علاج، يمكن أن يؤدي هذا المرض إلى الفشل الكبدي القاتل. هبوط سكر الدم، وزيادة بروتين يسمى AFP تحدث غالبا في الاطفال الرضع. في حالة عدم المعالجة، قد يحدث فشل في الكبد والكلى، وغالبا يموت الاطفال قبل سنة العشر سنوات بسبب فشل الكبد اوسرطان الكبد أو مضاغفات عصبية تسبب فشل في التنفس. . من العلامات المتكررة لدى هؤلاء المرضى هو ان رائحة البول تشبه الملفوف أو الزبدة المتعفنة.[5]
المعالجة
اتباع نظام غذائي صارم بحيث يكون الغذاء منخفض البروتين. أظهرت الأبحاث الحديثة أن الدواء الجديد الذي يسمى نيتيسينون (Nitisinone) أنه فعّال. نيتيسينون هو عقار مثبط لإنزيم يلعب دورا هاما في ايض التيروسين وهو العفار الوحيد حاليا المتوفر لعلاج فرط التيروزين الوراثي من نوع 1 مع التقييد بالحمية الغذائية. قد لا يستجيب 10% من المرضى لهذا العلاج[6] تعتبر زراعة الكبد العلاج النهائي والأكثر فعالية في المرضى الذين يعانون من نوع فرط تيروزين الدم من النوع 1.[7]
المراجع
- Reference, Genetics Home. "tyrosinemia". Genetics Home Reference (باللغة الإنجليزية). مؤرشف من الأصل في 28 يوليو 2018. اطلع عليه بتاريخ 27 أكتوبر 2017. الوسيط
|CitationClass=تم تجاهله (مساعدة) - De Braekeleer, M.; Larochelle, J. (August 1990). "Genetic epidemiology of hereditary tyrosinemia in Quebec and in Saguenay-Lac-St-Jean". American Journal of Human Genetics. 47 (2): 302–307. ISSN 0002-9297. PMID 2378355. مؤرشف من الأصل في 25 يناير 2018. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Gentz, J.; Jagenburg, R.; Zetterstroem, R. (April 1965). "TYROSINEMIA". The Journal of Pediatrics. 66: 670–696. ISSN 0022-3476. PMID 14271358. مؤرشف من الأصل في 25 يناير 2018. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Hostetter, M. K.; Levy, H. L.; Winter, H. S.; Knight, G. J.; Haddow, J. E. (1983-05-26). "Evidence for liver disease preceding amino acid abnormalities in hereditary tyrosinemia". The New England Journal of Medicine. 308 (21): 1265–1267. doi:10.1056/NEJM198305263082105. ISSN 0028-4793. PMID 6188953. مؤرشف من الأصل في 26 يناير 2018. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Enns GM, Packman S (2001). "Diagnosing Inborn Errors of Metabolism in the Newborn: Clinical Features" (PDF). NeoReviews. 2 (8): e183–e191. doi:10.1542/neo.2-8-e183. ISSN 1526-9906. اطلع عليه بتاريخ أكتوبر 2020. الوسيط
|CitationClass=تم تجاهله (مساعدة); تحقق من التاريخ في:|تاريخ الوصول=(مساعدة) - Holme, E.; Lindstedt, S. (August 1998). "Tyrosinaemia type I and NTBC (2-(2-nitro-4-trifluoromethylbenzoyl)-1,3-cyclohexanedione)". Journal of Inherited Metabolic Disease. 21 (5): 507–517. ISSN 0141-8955. PMID 9728331. مؤرشف من الأصل في 04 نوفمبر 2017. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Sokal, E. M.; Bustos, R.; Van Hoof, F.; Otte, J. B. (November 1992). "Liver transplantation for hereditary tyrosinemia--early transplantation following the patient's stabilization". Transplantation. 54 (5): 937–939. ISSN 0041-1337. PMID 1440864. مؤرشف من الأصل في 25 يناير 2018. الوسيط
|CitationClass=تم تجاهله (مساعدة)
 بوابة طب
بوابة طب