داء اختزان الغلايكوجين النمط الثاني
داء اختزان الغلايكوجين النمط الثاني (Pompe disease )
| Glycogen storage disease type II | |
|---|---|
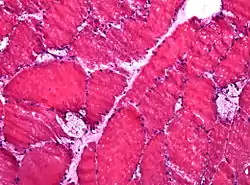 خزعة عضلية تظهر فجوات كبيرة في حالة داء بومبه (صبغة HE , مقطع مجمد). خزعة عضلية تظهر فجوات كبيرة في حالة داء بومبه (صبغة HE , مقطع مجمد). | |
| معلومات عامة | |
| الاختصاص | علم الغدد الصم |
| من أنواع | داء اختزان الغلايكوجين ، واعتلال عضلة القلب الضخامي |
| الإدارة | |
| أدوية | |
داء اختزان الغلايكوجين هو مرض وراثي جسمي متنحي أيضي,[1] والذي يسبب تلف في خلايا الأعصاب والعضلات وهو ناتج من طفره وراثيه في الجين (acid alpha-glucosidase (GAA مما يؤدي إلى تراكم الغلايكوجين في الحويصلة(1)(2) , وصف هذا المرض لأول مره 1932 على يد الطبيب جونس بومبي ومن هنا اكتسب المرض هذا الاسم فيدعى أحيانا بمرض بومبي . تراكم الغلايكوجين يسبب في ضعف العضلة ويؤثر على أغشية الجسم خاصة في القلب، الهيكل العضلي، الكبد والجهاز العصبي .
التصنيف
هناك استثناءات، ولكن مستويات ألفا جلوكوزيد يحدد نوع GSD الثاني عندالفرد. تراكم ألفا جلوكوسيديز موجودة في عضلات الأفراد يعني الاعراض التي تحدث في وقت لاحق في الحياة والتقدم ببطء أكثر. ينقسم GSD الثاني على نطاق واسع في شكلين الظهور اعتمادا على حدوث أعراض التقدم في السن.[2] وعادة ما يتم تشخيص الشكل عند الاطفال بالظهور في 4-8 أشهر. العضلات تبدو طبيعيه ولكن تكون في حالة ارتخاء وضعف تمنعهم من رفع رؤوسهم أو التدحرج .مع تقدم المرض عضلات القلب تتضخم وتفشل تدريجيا. دون العلاج عادة الموت يحدث نتيجة لفشل القلب وضعف في الجهاز التنفسي.[2] واحدة من الأعراض الأولى هو الانخفاض التدريجي في قوة العضلات بدءا من الساقين وتحريك العضلات الصغيرة في الجذع والذراعين، مثل الحجاب الحاجز والعضلات الأخرى المطلوبة للتنفس. فشل الجهاز التنفسي هي السبب الأكثر شيوعا للوفاة. توسيع أو تضخم عضلات القلب واضطرابات نبضات القلب ليست ميزات كبيرة ولكن لم تحدث في بعض الحالات.[2]
الأسباب
تخزين الجليكوجين نوع المرض الثاني له نمط وراثي جسمي متنحي. وهذا يعني ان الجين الذي يوجد فيه الخلل يقع على كروموسوم جسمي، ويوجد نسختين من الجين واحد من الاب والاخر من الام كما هو الحال مع جميع حالات الامراض الوراثيه، الأطفال لديهم فرصة 1 في 4 من وراثة هذا الاضطراب عندما يحمل كلا الوالدين الجينات المعيبة، وعلى الرغم من أن كلا الوالدين يحمل نسخة واحدة من الجينات المعيبة، فإنها عادة لا تتأثر الاضطراب. وينجم هذا المرض عن طفرة في جين (حمض ألفا جلوكوزيد: المعروف أيضا باسم حمض مالتاز) على الذراع الطويلة من كروموسوم 17 في 17q25.2-q25.3 (زوج قاعدة 75689876 إلى 75708272). عدد الطفرات وصفها حاليا (في عام 2010) 289 مع 67 يجري الطفرات غير المسببة للأمراض و 197 الطفرات المسببة للأمراض. لا يزال يجري تقييم ما تبقى لارتباطهم مع المرض. الجين حوالي 20 كيلو بايت ويحتوي على 20 الإكسونات مع واحد من الاكسون ليس لديه شيفره. محتوى GC مرتفعة (80٪) وسلسلة TATA متميزة وهناك نقص في العناصر CCAAT. ويبدو أن معظم الحالات تظهر نتيجة ثلاثة الطفرات. استبدال (T → G) هي الأكثر شيوعا بين البالغين الذين يعانون من هذا الاضطراب. هذه الطفرة تكون في موقع من الربط الحمض النووي الريبي. الجين يشفر ألفا جلوكوزيد البروتين الحمضية (EC 3.2.1.20) -الذي هو هيدرولاز الليزوزومية. البروتين هو انزيم أن يحط عادة ألفا -1,4 -1,6 وألفا الروابط في الجليكوجين، المالتوز وإيزومالتوز ومطلوب لتحلل 1-3٪ من الجليكوجين الخلوي. ونقص هذا الانزيم يؤدي إلى تراكم الجليكوجين العادي هيكليا في الجسيمات الحالة والسيتوبلازم في الأفراد المتضررين. تخزين الجليكوجين المفرط في الجسيمات الحالة قد يعوق الأداء الطبيعي للعضيات أخرى وتؤدي إلى الإصابة الخلوية. و تم التعرف على حمض ألفا المتعلقة جلوكوزيد الجين في الديدان الخيطية ايليجانس انواع معينة.
روابط خارجية
- The website of the Pompe's Group of the Association for Glycogen Storage Disease (UK)
- Acid Maltase Deficiency Association, Inc.—A US patient support group founded in 1995.
- United Pompe Foundation
- International Pompe Association—A federation of Pompe disease patient's groups worldwide.
- Genzyme's Pompe Information site
- FDA Approval News for Myozyme
- Canadian Association of Pompe
- Hide & Seek Foundation for Lysosomal Disease Research
- GeneReview/NIH/UW entry on Glycogen Storage Disease Type II (Pompe Disease)
- Australian Pompe's Association
- Asociación Española de Enfermos de Glucogenosis
- Fact files on Pompe Disease
- Understanding Pompe Disease - US National Institute of Arthritis and Musculoskeletal and Skin Diseases
مراجع
- Pompe disease على مرجع المكتبة الوطنية للطب الرئيسي لعلم الوراثة.
- "Type II Glycogen Storage Disease". The Association for Glycogen Storage Disease. مؤرشف من الأصل في 23 يناير 2013. اطلع عليه بتاريخ 22 مايو 2012. الوسيط
|CitationClass=تم تجاهله (مساعدة)
 بوابة طب
بوابة طب