متلازمة دوبويتز
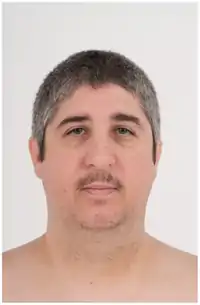
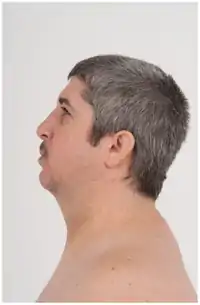
متلازمة دوبويتز هي اضطراب وراثي ونموي نادر يتميز بصغر الرأس، وتأخر النمو، ومظهر الوجه المميز (صغير، مستدير، ثلاثي الشكل مع ذقن مدبب، انحسار، أنف واسع، ذو حواف واسعة؛ عيون واسعة النطاق مع تدلى الجفون). الأعراض تختلف بين المرضى، ولكن تشمل الخصائص الأخرى: لينة، صوت ضارية عالية. حزام جزئي من الأصابع وأصابع القدمين. تشوهات الحنك. تشوهات الأعضاء التناسلية. التهاب الجلد. فرط النشاط؛ تفضيل التفكير الملموس على المجردة؛ الصعوبات اللغوية؛ والنفور من الحشود.[2] التسبب في المرض لم يتم بعد تحديدها، ولا الاختبارات الطبية يمكن تشخيص المرض بشكل قاطع.[3] الطريقة الرئيسية للتشخيص هو التعرف على المظاهر الوجهية. منذ أن وصفت لأول مرة في عام 1965 من قبل الطبيب الإنجليزي فيكتور دوبويتز، تم الإبلاغ عن أكثر من 140 حالة في جميع أنحاء العالم. على الرغم من أن معظم الحالات قد تم الإبلاغ عنها من الولايات المتحدة وألمانيا وروسيا، ويبدو أن هذا الاضطراب يؤثر على كلا الجنسين وجميع العرقيات على قدم المساواة.
متلازمة دوبويتز | |
|---|---|
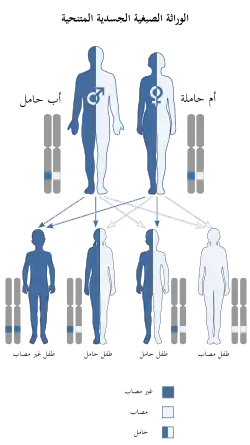 ورثت هذه الحالة عبر بطريقة مقهورة ورثت هذه الحالة عبر بطريقة مقهورة | |
| معلومات عامة | |
| الاختصاص | علم الوراثة الطبية |
| من أنواع | متلازمة ، وتدلي الجفن ، واضطراب صبغي جسدي متنحي |
| المظهر السريري | |
| الأعراض | تدلي الجفن [1] |
المظهر
صغر الرأس هو سمة فيها محيط الرأس أصغر من المعتاد بسبب التطور غير سليمة من الدماغ. هو سببه اضطرابات وراثية، التهابات، إشعاع، دواء أو إدمان كحول أثناء حالة حمل. العيوب في نمو القشرة المخية تؤدي إلى العديد من الميزات المرتبطة صغر الرأس.[4] لا توجد حاليا طريقة لعلاج هذا الشذوذ بطريقة من شأنها أن تعيد الرأس أو العقول إلى حجمها الطبيعي. ومع ذلك، هناك برامج العلاج التي هي أعراض وداعمة مثل العلاج البدني والكلام والأدوية للمضبوطات وفرط النشاط.[5] صغر الرأس لديه مجموعة واسعة من التكهن: بعض المرضى الذين يعانون القليل من التخلف العقلي جدا، ويمكن أن تصل إلى مراحل منتظمة العمر المناسب. البعض الآخر قد يعانون من التخلف العقلي الشديد والآثار الجانبية العصبية والعضلية.
وراثة المرض
على الرغم من أن علم الأمراض الدقيق لمتلازمة دوبويتز غير معروف حتى الآن، هو وراثي وتصنف على أنها مرض متنحية راثي. وعلاوة على ذلك، هناك قرابة بين الوالدين أحيانا. العديد من الحالات تشير إلى متلازمة دوبويتز التي تحدث في التوائم الزيجوت الواحدة، والأشقاء، وأبناء العم.[6] هناك تباين المظهري كبير بين الحالات،[7] وخاصة فيما يتعلق المخابرات. على الرغم من أدلة كبيرة تشير إلى الأساس الجيني لهذا الاضطراب، تم العثور على التشابه المظهري إنفيتال متلازمة الكحول. هناك حاجة إلى مزيد من الدراسات لتحديد ما إذا كان هذا العامل البيئي له تأثير على التعبير عن النمط الوراثي.[8] ومن المعروف أن انهيار الكروموسومات يحدث.
هرمون النمو
ويرافق متلازمة دوبويتز من نقص في هرمون النمو.[9] الأفراد الذين يعانون من هذا الاضطراب قد توقف النمو، وتفرز هرمونات النمو من قبل الغدة النخامية الأمامية من الدماغ. وتتمثل المهمة الرئيسية للنخامية الأمامية في زيادة ارتفاع الفرد أثناء التنمية. الغدة النخامية الأمامية تلعب أيضا دورا في تنظيم وظيفة المناعة، وزيادة الاحتفاظ بالكالسيوم، وزيادة كتلة العضلات وتحفيز غلوكونيوجينيسيس. نقص في هرمون النمو قد يكون سببه الطفرات الجينية، تشوهات في المهاد أو الغدة النخامية أثناء التنمية أو الأضرار التي لحقت الغدة النخامية.[10] في متلازمة دوبويتز، والسبب هو على الأرجح بسبب الطفرات الجينية أو تعطيل هياكل الدماغ خلال التنمية. نقص هرمون النمو يرتبط أيضا مع مستويات منخفضة من مفتش، وهي حالة وجدت في دوبويتز المرضى.
فيما يتعلق بـ SLOS
كما يحقق الباحثون في أوجه التشابه الجينية بين متلازمة دوبويتز ومتلازمة سميث-ليملي-أوبيتز (سلوس). المرضى الذين يعانون من سلوس ودوبويتز المتلازمات تجربة العديد من نفس التشوهات، وافتراضين اثنين من المفترض أن تكون مرتبطة. ومن سمات سلوس هو انخفاض مستوى الكوليسترول في الدم وارتفاع مستوى 7-ديهيدروكوليستيرول. الكولسترول ضروري لعدة وظائف رئيسية من الجسم، بما في ذلك بنية غشاء الخلية، وتوليد الجنين، والستيرويد وتركيب هرمون الجنس. انخفاض في نسبة الكوليسترول الحيوي أو النقل ربما يمثل معظم أعراض كل من سلوس ودوبويتز. على الرغم من أن عدد قليل فقط من المرضى الذين يعانون من متلازمة دوبويتز تم تحديدها مع مستويات الكولسترول المتغيرة، والباحثين استكشاف ما إذا كان دوبويتز متلازمة، مثل سلوس، يحمل صلة إلى خلل في مسار الحيوي الكوليسترول.[11]
لا يزال علم الأمراض البيوكيميائية الدقيق للمرض قيد البحث بسبب انخفاض معدل انتشار المرض ومجموعة واسعة من الأعراض المرتبطة به. وقد ركزت عدة دراسات على جوانب مختلفة من المرض في محاولة للعثور على سببها والتعبير الدقيق. دراسة واحدة فحص الميزات الشفوية محددة في مريض واحد.[12] وجدت أخرى تشوهات في الدماغ، مثل خلل خلل الجسم الثفني، وهو الغدة النخامية الأمامية المتطورة وساق الدماغ مع نيوروهيبوفيسيس خارج الرحم.[13]
مراجع
- معرف أنطولوجية المرض: http://www.disease-ontology.org/?id=DOID:14796 — تاريخ الاطلاع: 29 نوفمبر 2020 — الرخصة: CC0
- "Dubowitz syndrome". Encyclopedia of Genetic Disorders. مؤرشف من الأصل في 18 أبريل 2007. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Dubowitz Syndrome Support Network". مؤرشف من الأصل في 9 مايو 2008. الوسيط
|CitationClass=تم تجاهله (مساعدة) - Microcephaly Information Page على موقع المعهد الوطني للاضطرابات العصبية والسكتة الدماغية (NINDS)
- "Microcephaly - Symptoms, Treatment and Prevention". The HealthCentralNetwork. مؤرشف من الأصل في 6 فبراير 2012. الوسيط
|CitationClass=تم تجاهله (مساعدة) - الوراثة المندلية البشرية عبر الإنترنت (OMIM) Rasmussen, Sonja A. Dubowitz Syndrome -223370
- "Dubowitz syndrome: possible evidence for a clinical subtype". Am. J. Med. Genet. 35 (4): 561–5. 1990. doi:10.1002/ajmg.1320350423. PMID 2185633. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "[Dubowitz syndrome. A diagnosis not to be missed]". Arch. Fr. Pediatr. (باللغة الفرنسية). 48 (10): 715–8. December 1991. PMID 1793348. الوسيط
|CitationClass=تم تجاهله (مساعدة) إخلاء مسؤولية طبية
إخلاء مسؤولية طبية بوابة طب
بوابة طب
- "Growth hormone deficiency in Dubowitz syndrome". Acta Paediatr Jpn. 38 (3): 267–9. 1996. doi:10.1111/j.1442-200x.1996.tb03484.x. PMID 8741320. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Archived copy". مؤرشف من الأصل في 24 أبريل 2007. اطلع عليه بتاريخ 30 أبريل 2007. الوسيط
|CitationClass=تم تجاهله (مساعدة)صيانة CS1: الأرشيف كعنوان (link) Rieser, Patricia. Growth Hormone Deficiency. 1979. 30 April 2007. - "Dubowitz syndrome: a defect in the cholesterol biosynthetic pathway?". Am. J. Med. Genet. 86 (5): 503–4. 1999. doi:10.1002/(SICI)1096-8628(19991029)86:5<503::AID-AJMG21>3.0.CO;2-Y. PMID 10508998. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Dubowitz syndrome: report of a case with emphasis on the oral features". J Dent Child (Chic). 72 (3): 100–3. 2005. PMID 16568913. مؤرشف من الأصل في 13 ديسمبر 2019. الوسيط
|CitationClass=تم تجاهله (مساعدة) - "Cranial midline abnormalities in Dubowitz syndrome: MR imaging findings". Eur Radiol. 13 (5): 1056–7. 2003. doi:10.1007/s00330-002-1580-2. PMID 12695828. الوسيط
|CitationClass=تم تجاهله (مساعدة)